Chapter Outline
SEVERITY OF RH HEMOLYTIC DISEASE
PREDICTION OF FETAL HEMOLYTIC DISEASE
MANAGEMENT OF MATERNAL ALLOIMMUNIZATION
MANAGEMENT OF SPECIAL PROBLEMS
OTHER MATERNAL ALLOANTIBODIES CAUSING FETAL AND NEONATAL HEMOLYTIC DISEASE
Historical Aspects
Whereas many serious complications of pregnancy were well known in antiquity, maternofetal blood group incompatibility was recognized more recently. Against the high perinatal mortality prevailing until the mid-twentieth century, deaths from hemolytic disease were of little statistical significance, even though about half of the affected babies died.
In 1932 Diamond and coworkers deduced the connection between “congenital anemia,” icterus gravis, and hydrops fetalis, and coined the term erythroblastosis fetalis for the disease characterized by hemolytic anemia, intramedullary and extramedullary erythropoiesis, and hepatosplenomegaly. Darrow correctly speculated in 1938 that the disease was caused by maternal antibodies to fetal antigens developed as a result of transplacental fetomaternal hemorrhage (FMH).
In 1939 Levine and Stetson linked a woman’s severe transfusion reaction to her husband’s blood with her recent delivery of a hydropic stillborn infant. She was found to have an antibody that agglutinated her husband’s red blood cells (RBCs). Levine postulated that she had become sensitized to an antigen that the fetus had inherited from its father.
In 1940 Landsteiner and Wiener proposed the identity of the antigen by generating antibodies to rhesus monkey RBCs in guinea pigs and rabbits. The antisera agglutinated RBCs from 85% of white people. These people were designated rhesus (Rh) positive. The 15% whose RBCs were not agglutinated were rhesus (Rh) negative.
Subsequent years saw rapid and dramatic progress, including the development of the Coombs test. This test uses anti–human immunoglobulin G (IgG) antibodies to agglutinate IgG-labeled RBCs and remains important in the detection and management of hemolytic disease of the fetus and newborn (HDFN). Chown reported in 1954 that mothers could be sensitized by a transplacental hemorrhage of fetal blood, although until crossmatching for Rh groups became routine, transfusion for postpartum hemorrhage and other indications also contributed to the incidence of the disease. Chown’s observation and the subsequent recognition that most sensitizing FMHs occurred at delivery, explaining why Rh HDFN was mainly a disease of multipara, paved the way for development of immunoprophylaxis. Since the late 1960s RhD immunoglobulin has been available to treat RhD-negative mothers of RhD-positive infants at delivery to prevent Rh HDFN.
For many problems of pregnancy, obscurity of the pathophysiology often led to management that, until recently, was symptomatic at best. In contrast, with hemolytic disease the genetic predilection, molecular mechanism, and rationale for treatment were rapidly deduced, which led to remarkable success in preventing the disease and mitigating its effects. Indeed, the history of the prevention and management of RhD HDFN includes a number of pivotal discoveries that have influenced the management of other diseases. These breakthroughs include exchange transfusion for neonatal treatment, development of screening programs for maternal blood group antibodies, strategies for managing early delivery to optimize neonatal outcomes, the use of amniocentesis for fetal diagnosis, the first example of intrauterine fetal treatment, the development of one of the earliest and most successful forms of immunoprophylaxis using human antibodies, and the noninvasive prediction of fetal RHD type using DNA from maternal blood. Subsequently, many of the successes of fetal and neonatal treatment of Rh HDFN have been extrapolated to HDFN caused by other blood group antibodies and other problems of pregnancy.
Advances in molecular biology and biochemistry allowed the identification of the Rh polypeptides in 1982 and the RH genes in 1991. These discoveries amended previous suggested models to explain inheritance, although like many disorders that were once thought to have been genetically simple and phenotypically diverse, the diversity of Rh genotype/phenotype relationships is much greater than once imagined.
The Rh Blood Group System
Biochemistry and Molecular Genetics
Although the incidence of Rh HDFN has declined and the incidence of HDFN caused by other alloantibodies has increased, the Rh blood group system remains the most common cause of HDFN (particularly severe HDFN), especially in white persons, for whom approximately one in seven conceptions involves an Rh-negative woman and an Rh-positive man. Individuals are classed as Rh positive or negative based on expression of the major D antigen on their RBCs; however, more than 50 other Rh antigens have been identified, of which the most commonly recognized in clinical practice are those designated D, C, c, E, and e. The Rh blood group system is the most complex blood group system in humans.
After the landmark descriptions of the RhD protein in 1982, the non-D and D complementary DNAs (cDNAs) and genes were cloned in the early 1990s. In most individuals the Rh blood group loci are the products of two very similar genes, one of which encodes the polypeptide that carries the Rh CcEe epitopes in combinations (Ce, ce, cE, or CE); the other encodes the RhD antigen polypeptide ( Table 3-1 ). The two genes are separated by only 30 kb (including the small SMP1 gene) and are organized in opposite orientation on chromosome bands 1p36.1-p34.3. They share a very similar 10-exon structure, and their intronic sequences are also highly conserved. Their sequence identity strongly suggests that they evolved by duplication of an ancestral gene approximately 8 million years ago. Worldwide, the Rh blood group system displays considerable polymorphism. A wide variety of both small and large mutations (including gene deletion and recombination to form hybrid RHD-RHCE products) and the effects of other modifying genes account for the diverse common and rare Rh phenotypes. Although some genotypic and phenotypic variants are confined to a single family, for others the incidence in various gene pools is much higher or unknown. This diversity mandates caution in the use of DNA-based clinical diagnostic approaches, especially when these approaches are applied to populations other than the ones in which they were piloted, but these techniques may open the door for future individualization of prevention and therapy.
| Gene | Allele | Translated Product | Allele Frequency in United States (%) | |||
|---|---|---|---|---|---|---|
| Whites | Blacks | Native Americans | Asians | |||
| RHD | RHD | RhD polypeptide | 61 | 97 | 99 | 99 |
| RHD deletion or silent | None | 39 | 3 | 1 | 1 | |
| RHCE | RHCe | RhCe polypeptide | 44 | 19 | 46 | 72 |
| RHcE | RhcE polypeptide | 15 | 11 | 40 | 21 | |
| RHce | Rhce polypeptide | 41 | 70 | 8 | 6 | |
| RHCE | RhCE polypeptide | 0 | 0 | 6 | 1 | |
It has been well established for decades that no d antigen exists. RhD negativity is defined as the absence of RhD antigen, and its incidence is highest in the Basque population. It has been suggested that peripatetic Basque traders and fishermen (or their ancestors) may have spread this genotype to other populations, which explains its higher prevalence in those of European ancestry ( Table 3-2 ). In European-descended populations in which RhD negativity is relatively common, most RhD-negative individuals are homozygous for an RHD gene deletion. This mutation appears to have arisen because two highly homologous sequences (1463 bp “rhesus boxes”) flank the RHD gene and provide the opportunity for unequal crossing over. However, although RhD negativity is less common among them, other mechanisms for D negativity appear more commonly in other populations, such as those of African, Chinese, or Japanese ancestry. These alternative mechanisms include partial deletion, recombination between RHD and RHCE genes, and point mutation. An Rh allele that comprises a pseudogene ( RHDψ ) is prevalent among RhD-negative people of African ancestry, including approximately 66% in South Africa and about one in five RhD-negative African Americans. RHDψ is not expressed because of two inactivating mutations. Singleton found that 15% of RhD-negative black South Africans carry a hybrid RHD-CE-D that also encodes an altered C antigen.
| Nomenclature | Expressed Alleles | Frequency | ||||
|---|---|---|---|---|---|---|
| Fisher-Race | Wiener | Whites | Blacks | Asians * | ||
| RhD positive | CDe | R 1 | RHD RHCe | 0.42 | 0.17 | 0.70 |
| cDE | R 2 | RHD RHcE | 0.14 | 0.11 | 0.21 | |
| cDe | R 0 | RHD RHce | 0.04 | 0.44 | 0.03 | |
| CDE | R z | RHD RHCE | 0.00 | 0.00 | 0.01 | |
| TOTAL | 0.6 | 0.72 | 0.95 | |||
| RhD negative † | cde | r | RHce | 0.37 | 0.26 | 0.03 |
| Cde | r’ | RHCe | 0.02 | 0.02 | 0.02 | |
| cdE | r” | RHcE | 0.01 | <0.01 | <0.01 | |
| CdE | r y | RHCE | <0.01 | <0.01 | <0.01 | |
| TOTAL | 0.4 | 0.28 | 0.05 | |||
| Ratio of RhD-positive to RhD-negative phenotypes | 0.84 : 0.16 | 0.92 : 0.08 | 0.99:<0.01 | |||
* The predominant reasons for failure to express RHD vary by racial group. RHD deletions predominate in whites, but other variants, such as partial deletions and RHD-RHCE recombinations, are more common in other populations.
The molecular basis of the RhD-positive phenotype is also intricate. Studies using site-directed mutagenesis, naturally occurring variants of RHD, and monoclonal antibodies or molecular modelling indicate that the RhD antigen is composed of at least 40 different but overlapping epitopes, some of which are missing in individuals who express partial D antigenicity. The RHD and RHCE gene products are unique among blood group antigens in being nonglycosylated, palmitoylated integral membrane proteins ( Fig. 3-1 ). Their expression is confined to erythroid cells, whereas many other blood group antigens are present in many tissues.
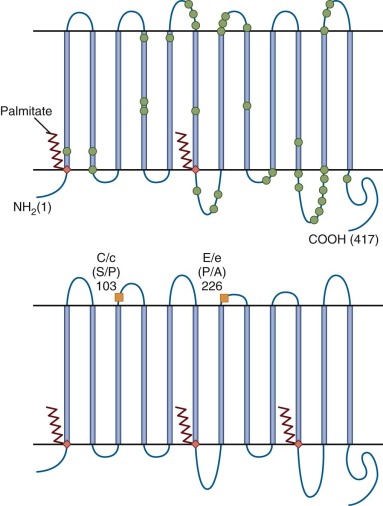
The RhD and RhCE antigens are found in association with a glycoprotein, formerly known as RH50 glycoprotein and now designated Rh-associated glycoprotein (RhAG). RHAG shares about one third homology with RHD and CE ; however, it maps to 6p21.1-p11. The Rh complex comprises multimers of RhD or CE subunits with RhAg.
Cartron and Cartron and Colin have reviewed the Rh deficiency states. Rare individuals with the Rh deficiency syndrome (Rh null ) lack expression of all or virtually all RhD, C/c, and E/e antigens. This recessive condition can result (in RhD-negative individuals) from homozygous silent mutations in the RHCE gene that appear to reduce cell surface expression of RhAG (“amorph type”). Homozygotes for mutations that prevent expression of RhAG have the more common “regulator type” or the Rh mod phenotype. Despite normal transcription of RH genes, the proteins are not found at the cell surface.
The functional significance of the Rh complex is indicated by the fact that Rh-deficient individuals have spherocytosis, stomatocytosis, and chronic hemolysis of varying severity. Their RBCs have altered ion transport and membrane composition and are partly or completely deficient in certain other membrane proteins including RhAG, CD47, LW (Landsteiner-Wiener glycoprotein, or intracellular adhesion molecule 4 [ICAM-4]), GPB (glycophorin B, which confers the Ss blood group), and Duffy Fy5 (Duffy antigen receptor for chemokines). These deficiencies suggest that Rh proteins form complexes with RhAG and other proteins and that the complexes fail to form if components are missing (see Chapter 15 ). There is also strong evidence for an interaction with the anion-exchange protein (band 3) and linkage of the Rh complex to the spectrin-based RBC membrane skeleton, thus indicating a role in maintaining the biconcave disc shape of RBCs (see Fig. 15-6 in Chapter 15 ). Failure of this association may explain some of the dysfunction of Rh-deficient cells. However, many details of when, how, and why the Rh complexes form remain to be explained.
In contrast to the common Rh-negative and very rare Rh deficiency states, weak and partial D phenotypes are fairly common (e.g., 0.2% to 1% of populations of European ancestry) and are caused by RHD coding region alterations that affect the number and antibody binding characteristics of RhD sites per cell. The term “partial D” has been used to describe RBCs that react with some monoclonal anti-D antibodies and not others, and their RhD proteins typically have one or more amino acid substitutions on or near the RBC surface, or consist of RhD-RhCE hybrids. In contrast, “weak D” has been used for RBCs with markedly reduced numbers of antigenic sites and in which one or more amino acid substitutions are found in regions that are presumed to be in or below the membrane and may interfere with assembly of Rh complexes. Most individuals with weak D, especially those with types 1, 2, and 3, do not make anti-D. However, it has been recognized that there is overlap in, and some imprecision in allocation of, these phenotypes, and the terms “aberrant D” or “D variant” have been proposed as collective terms for these plus the (very weak) DEL phenotype ( Table 3-3 ).
| Category | Phenotype | Typical Mutations |
|---|---|---|
| Partial D | D II to D VII and others | Variety of point mutations and RHD-RHCE hybrids |
| Weak D | Weak D (D u ) types 1-16 | Variety of point mutations leading to amino acid substitutions in transmembrane or cytoplasmic domains |
| D negative (silent D or d) | D negative | Variety of mechanisms: RHD complete or partial deletion, CE hybrids, point mutations; some prevent transcription, but all prevent expression at the cell surface |
| CE variants | RN, E I-III , VN, CX, C W , etc. | Variety of hybrids and point mutations |
| CE silent | D.., D-, and various others | Mostly RHD-RHCE hybrids |
| Rh deficiency states (no D or CE expression) | D null amorph | RHCE silent genes plus RHD deletion |
| D null reg , D null mod | RHAG mutations that prevent expression |
Because of variable antigenic potential and the potential to form antibodies to RhD, certain weak and partial D phenotypes are significant in transfusion therapy and pregnancy. Some individuals are considered RhD-negative in pregnancy and as transfusion recipients (eligible for anti-RhD immunoprophylaxis and Rh-negative blood) but Rh-positive as blood donors. For example, if some blood donors with aberrant, or variant, D are regarded as D negative, their blood can cause alloimmunization of D-negative recipients. On some occasions, women with D variants (especially partial D VI) develop anti-D antibodies capable of causing severe HDFN if they carry a D-positive child, or D-negative women can develop antibodies if their babies have some types of aberrant, or variant, D. Some partial D phenotypes are also associated with distinct antigens.
The C antigen is most common in southeast Asia, parts of South America, and the southern tip of Greenland, whereas the E antigen occurs most often in populations indigenous to parts of South America and Alaska. The genetics underlying RhCc/Ee phenotypes also display more subtle variation than was once envisaged, and weak, partial, and variant antigen phenotypes have also been recognized. Internet databases are a good source of up-to-date information about the phenotype/genotype relationships for RhD and RhCE.
RhD Alloimmunization
HDFN is a disease of the fetus caused by a maternal response to pregnancy. As such, the pathogenesis can be considered in three stages: maternal alloimmunization, antibody transfer to the fetus, and fetal response.
Maternal Rh Sensitization
Maternal sensitization to Rh antigens can occur as a result of exposure to antigenically dissimilar fetal RBCs during pregnancy, as a result of therapeutic transfusion, or occasionally as a result of needle sharing. In high-resource countries, transfusion rarely causes RhD sensititization because compatibility testing for RhD antigen has been a routine practice for several decades. However, it accounts for a high proportion of sensitization to other blood group antigens, including RhCE, and among those with RhD and RhCE variants. Furthermore, the need for emergency transfusion of Rh-positive blood to Rh-negative women is likely to remain a cause of sensitization to RhD in locations where the Rh-negative phenotype is rare and Rh-negative blood is scarce.
Exposure to fetal antigens remains the most common cause of maternal antibodies to RhD. Although Chown first described maternal Rh immunization that occurred as a result of FMH, the test of Kleihauer and associates in 1957 was needed to show the incidence, size, and timing of these events and to show that most maternal Rh sensitization occurs this way. The test, which depends on the resistance of fetal hemoglobin (HbF) to acid elution ( Fig. 3-2 ), can detect 1 fetal RBC in 200,000 adult RBCs, corresponding to a fraction of a milliliter in the circulating maternal blood volume. However, acid elution studies must be interpreted with some caution because of rapid destruction of fetal cells when ABO incompatibility exists and natural variation and pregnancy-induced increases in expression of HbF among adults. Using flow cytometric techniques and DNA amplification of fetal sequences, researchers have subsequently confirmed that some fetal cells can be detected in maternal blood during all trimesters of most pregnancies. Because the strategies for detection vary among studies, there are inaccuracies of the Kleihauer-Betke test and there are uncertainties about the persistence of fetal cells in the maternal circulation, the volume and timing of this transplacental passage of fetal cells remain unclear. However, most of these “hemorrhages” are likely to be small in volume. Delivery is the time of greatest risk for FMH and larger hemorrhages, especially when complications of delivery of the fetus or placenta occur.
Larger FMHs are uncommon; the amount of fetal blood has been estimated to exceed 2.5 mL in fewer than 1% of gestations and is greater than 30 mL in fewer than 0.25%. However, certain obstetric situations increase the risk of significant FMH; these include antepartum hemorrhage, placental abruption, preeclampsia, external cephalic version, cesarean section, and manual removal of the placenta. Abdominal trauma and placental vascular malformations are additional but uncommon causes. FMH occurs after early-gestation chorionic villus sampling (CVS) and after amniocentesis, although the risk of maternal sensitization after amniocentesis is typically higher, perhaps because CVS is usually performed in earlier gestation when there is lower expression of fetal blood group antigens. The level of risk of sensitization as a result of first trimester pregnancy loss is also uncertain and in the case of spontaneous miscarriage will depend on whether an embryo with circulating blood ever formed, but surgical evacuation of the uterus is thought to present a greater risk of sensitization than medically induced or spontaneous evacuation. The Rh antigen may be expressed by approximately 38 days after conception, early enough in gestation for early CVS or pregnancy loss to present a potential risk.
All studies have focused on fetal cells in the maternal bloodstream, but the responses to submucosal, subcutaneous, or intramuscular antigen presentation differ from the responses to intravascular presentation. Therefore deposition of fetal RBCs in these sites could be more immunogenic than with the intravascular route and could account for some of the propensity for Rh sensitization after invasive procedures. If this is the case, techniques such as those recommended to minimize exposure of the fetus to maternal blood might be effective in reducing risk of blood group sensitization.
The Nature of Rh Sensitization
Primary and Secondary Immune Responses
In most RhD-negative individuals, the primary immune response develops slowly. In experimental Rh immunization of male volunteers, antibody responses are typically detected at 8 to 9 weeks, although they can occur at any time from 4 weeks to 6 months. Hemolytic anemia caused by anti-RhD has been detected as early as 10 days after a massive Rh-incompatible transfusion in a previously unsensitized patient. A primary, often weak immunoglobulin M (IgM) response will not affect the fetus even if it occurs before birth in a sensitizing pregnancy, because IgM anti-RhD does not cross the placenta. However, immunoglobulin G (IgG) anti-RhD production usually ensues, and if sensitization occurs in early pregnancy there is the potential for continued antigenic challenge, accounting for the rare occurrence of HDFN in the progeny of women during their first Rh-positive pregnancy.
After a primary response has been invoked, subsequent exposure to Rh-positive RBCs induces a rapid increase in anti-RhD IgG, which can cross the placenta and affect the fetus. Repeated exposures can progressively increase the antibody titer and change the characteristics (including IgG subclass distribution) of the RhD antibody, thus increasing the severity of Rh erythroblastosis in successive pregnancies.
Dose of Antigen Necessary to Produce RhD Sensitization
Experiments using injection of RhD-positive blood into RhD-negative male subjects show, not unexpectedly, that the likelihood of Rh sensitization depends on both dose and repetition of exposure to the antigen. A proportion of individuals are anergic to RhD regardless of dose and repetition of exposure (“nonresponders”).
Studies during and immediately after pregnancy using the Kleihauer technique indicated that if the volume of FMH is always less than 0.1 mL of RBCs, the prevalence of RhD sensitization detectable up to 6 months after delivery was 3%, whereas when volumes exceed 0.4 mL, the prevalence was 22%. Nevertheless, because in 75% to 80% of pregnancies the amount of the FMH is always less than 0.1 mL, it is likely that either small or undetectable FMHs or fetal cells deposited in other sites account for the majority of sensitizations. Secondary immune responses may occur after exposure to very small amounts of RhD-positive RBCs (as little as 0.03 mL).
Incidence of RhD Sensitization
RhD sensitization has decreased markedly with the introduction of RhD immunoprophylaxis. In its absence, about 16% of RhD-negative women become sensitized in their first ABO-compatible RhD-positive pregnancy. Of these, approximately one half have detectable anti-D 6 months after delivery, and in one half a rapid secondary response is seen in the next susceptible pregnancy, indicating that previous primary sensitization had occurred. If sensitization does not occur, the risk in a second RhD-positive, ABO-compatible pregnancy is similar. By the time an RhD-negative woman has completed her fifth ABO-compatible, RhD-positive pregnancy, the probability that she will be RhD sensitized is approximately 50%.
As parity increases and the number of women capable of an RhD immune response diminishes because they have already become immunized, the proportion of the remainder whose systems mount a primary immune response decreases because of a greater residual number of nonresponders. About 25% to 30% of RhD-negative women are nonresponders, in that they do not become RhD sensitized despite having many RhD-positive pregnancies; however, some may yet become RhD sensitized if they are exposed to a very large amount of RhD-positive blood. Immunologic tolerance can be induced by several different mechanisms and can depend on the context of antigen presentation to the immune system. It is not known which mechanisms of tolerance predominate in nonresponding RhD-negative women.
ABO incompatibility provides partial protection against RhD sensitization. Without anti-RhD immunoprophylaxis, it has been estimated that in white persons blood group A incompatibility between mother and fetus reduces the risk of RhD sensitization by 90% and group B by 55%. Bowman suggested that this partial protection is due to rapid intravascular hemolysis of the ABO-incompatible, RhD-positive RBCs, with sequestration of RhD-positive cells in the stroma in the liver (an organ with poor antibody-forming potential) rather than in the spleen (the site of RBC sequestration when extravascular RBC destruction occurs). However, it is not clear whether the immune system remains naïve with respect to the RhD antigens or tolerance is induced in these instances. Although ABO incompatibility provides substantial protection against the primary RhD immune response, it provides no protection against the secondary RhD immune response and should not influence management of an already affected pregnancy. Maternal HLA type does not have any consistent effect on risk of sensitization, although it may affect antibody titre and severity of HDFN.
Other factors that influence the likelihood and severity of sensitization include fetal gender (male to female, 1.44-1.74 to 1) and fetal red cell phenotype, which affects the number and antigenicity of RhD antigen sites on fetal RBCs. For example, R1r (CDe/cde) cells, with only 9900 to 14,400 D antigen sites per cell, are less immunogenic than R2r (cDE/cde) cells, which have 14,000 to 16,000 sites. The possibility that some RhD-negative women are exposed to RhD during their own fetal life (the so-called grandmother theory) has been considered, but the evidence from a case-control study is against it. Nevertheless, convincing evidence of maternal-to-fetal hemorrhage has been found in other circumstances, and these events could explain the occasional presence of RhD antibodies in RhD-negative men who have never had a transfusion.
RhD sensitization during pregnancy occurs in about 1.5% of susceptible pregnancies. The proportion of all occurrences of RhD sensitization that are attributable to antenatal sensitization now varies among studies, depending on the application of antenatal immunoprophylaxis.
In summary, some general rules apply to RhD HDFN. The probability of incompatible pregnancies is predictable from Hardy-Weinberg genetic principles, but the impact of HDFN on a population will also depend on typical family size and on careful management of obstetric complications. The risk should be low when good blood bank procedures, parsimonious approaches to blood transfusion, effective screening of pregnant women, and evidence-based administration of RhD immunoprophylaxis are applied. The disease should be uncommon or mild in a woman’s first incompatible pregnancy, and 50% of the infants of heterozygous fathers and sensitized mothers should be safe. However, the striking feature of pregnancies in severely isoimmunized women is their clinical variability. Because these women have often become sensitized despite the predictions of conventional wisdom, their care requires astute and conscientious management to ensure satisfactory outcomes.
Prevention of Rh Sensitization
The background to the prevention of RhD sensitization lies in the experiments of Emil von Dungern at the beginning of the twentieth century. He showed that serum from rabbits that had previously been injected with ox cells prevented the development of antibodies to ox cells in a second group of rabbits. The partial protective effect of ABO incompatibility also suggested that antibody-mediated destruction of fetal RBCs could prevent maternal RhD sensitization. Because anti-RhD immunoglobulin was strikingly successful in preventing sensitization in males, clinical trials were then undertaken in which RhD-negative, unsensitized women were given anti-D intramuscularly after delivery of an RhD-positive infant. The licensing of RhD immunoglobulin in 1968 profoundly influenced the prevalence of Rh sensitization. Despite its success, the exact mechanism of action of prophylactic RhD immunoglobulin is not yet fully elucidated.
The level of evidence for the use of postpartum RhD immunoglobulin is high. A systematic review collated data from the six eligible trials involving more than 10,000 women that compared postpartum RhD immunoglobulin with no treatment or placebo. The conclusion was that RhD immunoglobulin strikingly lowered the incidence of RhD alloimmunization 6 months after birth (relative risk 0.04, 95% confidence interval 0.02 to 0.06) and in a subsequent pregnancy (relative risk 0.12, 95% confidence interval 0.07 to 0.23). These benefits were seen regardless of the ABO status of the mother and baby when RhD immnoglobulin was given within 72 hours of birth. Higher doses (up to 200 µg) were more effective than lower doses (up to 50 µg) in preventing RhD alloimmunization in a subsequent pregnancy. The usual dose after full-term delivery in the United States is 300 µg (1500 International Units), which will effectively suppress the immunizing potential of approximately 17 mL of RhD RBCs (approximately 30 mL of fetal blood), although other guidelines recommend lower standard doses. Routine assessment of the size of FMH and subsequent titration of RhD immunoglobulin dose is recommended because although larger volume FMH is uncommon, it is not reliably predicted by clinical history. Postpartum prophylaxis is recommended whenever an RhD-positive infant is born to an unsensitized RhD-negative mother. RhD immunoglobulin is also recommended if the infant is positive for DVI or weak D. After postnatal immunoprophylaxis, the risk of RhD alloimmunization is between 1% and 2%. Administration of 100 µg (500 IU) of RhD immunoglobulin at 28 and 34 weeks’ gestation to women in their first pregnancy can reduce this risk to less than approximately 0.2%. Most current guidelines therefore recommend routine administration of RhD immunoglobulin at doses of 250 to 300 µg (1250-1500 IU) at about 28 weeks or 100 to 125 µg (500-750 IU) at 28 and 34 weeks’ gestation to all RhD-negative pregnant women.
Some transfusion services also recommend prophylaxis to weak D-positive women, although it is not recommended in most guidelines. Additional or earlier doses (depending on the timing and circumstances) for RhD-negative women who are not already sensitized and who experience trauma or who are undergoing amniocentesis, CVS, or other relevant procedures may further reduce the risk of sensitization. There is no high-quality evidence for immunoprophylaxis after early pregnancy loss or termination, but RhD immunoglobulin (typically in smaller doses) is widely recommended.
Small amounts of the RhD immunoglobulin cross the placenta and cause a weakly positive antiglobulin test, but there are no apparent adverse effects on the infant. It is important to note that after antenatal RhD immunoglobulin, results of maternal antibody screening tests done near term may be positive (albeit at low titer), but under the current guidelines these results should not preclude routine estimation of peripartum FMH and administration of postnatal immunoprophylaxis.
Approximately 1 in 400 women will have a FMH of greater than 30 mL of fetal blood at the time of delivery. Such hemorrhages are often clinically silent but can be detected by routine performance of a Kleihauer-Betke test on RhD-negative mothers of RhD-positive neonates. Prevention of RhD sensitization in the presence of such a large dose of antigen requires prompt assessment (by the Kleihauer-Betke test, preferrably confirmed by flow cytometry, which is more accurate but less widely available ) and administration of a titrated dose of RhD immunoglobulin. Pediatricians must be aware of this possibility and promptly notify their obstetric colleagues of any suspected FMH in the newborn infant of an RhD-negative mother. Assessment of the magnitude of FMH is also recommended after pregnancy complications such as antepartum hemorrhage and abdominal trauma.
Certain knowledge of RhD negativity in the biologic father of the fetus can obviate the need for antenatal immunoprophylaxis. However, this information should be established with a very high degree of certainty because of the potential for severe consequences in a future pregnancy in case of error.
Reports from numerous countries document the reduction in RhD HDFN as a result of RhD immunoglobulin. Nevertheless, there is no room for complacency because 30% to 40% of the reduction in incidence of HDFN over recent decades has been attributed to decreased family size rather than to immunoprophylaxis. Chavez and colleagues noted that the incidence of RhD sensitization in the United States is three times the rate predicted if RhD immunoglobulin was always applied according to guidelines issued by the American College of Obstetricians and Gynecologists. The failure of the immunoprophylaxis program to completely abolish RhD HDFN can be partly attributed to noncompliance with guidelines. In addition, recommendations for additional doses during pregnancy have led to scarcity of RhD immunoglobulin in some locations. These problems are even greater in low-resource countries. Giving booster doses to donors, using carefully screened RhD-positive RBCs, has been a necessity as the number of suitable sensitized donors decreases, but this practice requires extreme care to protect both the donor and eventual recipients. The distribution of the RhD-negative phenotype may increase as a result of migration. As with use of any blood product, issues of safety cannot be disregarded. All these forces threaten the supply of RhD immunoglobulin. Recombinant monoclonal or polyclonal preparations—which could, in principle, replace the current polyclonal RhD immunoglobulin, thereby resolving problems of availability and safety—have been under development in recent years. However, although monoclonal antibodies have significant diagnostic and research applications, they are not yet available for prophylaxis against sensitization.
Pathogenesis of Rh Hemolytic Disease
Erythropoiesis begins in the yolk sac by 14 days’ gestation, and Rh antigen is expressed by the sixth week. By 6 to 8 weeks’ gestation, the liver replaces the yolk sac as the main site of RBC production. Normally, erythropoiesis then diminishes in the liver and takes place nearly entirely in the bone marrow by the late third trimester of pregnancy.
Erythroblastosis is caused by maternal IgG coating of fetal RBCs, leading to their destruction. The subsequent fetal anemia stimulates erythropoiesis via the production of erythropoietin and other erythroid growth factors. When fetal marrow RBC production cannot keep up with RBC destruction, marked extramedullary erythropoiesis can be found in other organs, including the liver and in more severe cases also in the spleen, kidneys, skin, intestine, and adrenal glands. Hepatosplenomegaly is a hallmark of erythroblastosis fetalis. In the presence of erythroblastosis, immature nucleated RBCs, from normoblasts to early erythroblasts, are poured into the circulation.
Severity of Rh Hemolytic Disease
Degrees of Severity
For purposes of audit, research, and planning of treatment, degrees of severity of HDFN have been used since the 1950s.
In general, fetuses and newborns with mild HDFN have had antibody-coated RBCs, yielding positive results on a direct antiglobulin (Coombs) test, but they have no anemia or anemia that is well-compensated for in utero and after birth and do not require exchange transfusion to prevent bilirubin toxicity. This group historically accounted for approximately one half of affected fetuses. Nearly all survive and do well without invasive treatment.
Moderately affected fetuses are at risk of neural toxicity from bilirubin if they do not receive treatment (see Chapter 4 ). Historically, they are a group who routinely underwent exchange transfusion after birth. They are likely to have some signs of anemia in utero and ex utero but are not significantly acidotic or hydropic. Peripheral blood typically shows polychromasia, anisocytosis, and reticulocytosis, although these can be suppressed by intrauterine treatment ( Fig. 3-3 ). Maternal clearance copes with the products of hemolysis, and the fetus is usually born in good condition at or near term. This category historically represented approximately one third of fetuses. Moderately affected neonates do require vigilant neonatal management, including intensive phototherapy or exchange transfusion to manage jaundice ( Fig. 3-4 ).
Severely affected fetuses have hydrops or hydrops is impending, and they would die before, during, or after birth unless managed intensively. Untreated, hydrops develops in half between 18 and 34 weeks’ gestation and the other half between 34 and 40 weeks’ gestation. Polyhydramnios usually occurs early in the course of hydrops and results from decreased swallowing of amniotic fluid. Ascites is usually the first feature to develop, with severe anemia followed by skin edema and pleural and pericardial effusions. In infants with the most extreme conditions, compression hypoplasia of the lungs makes gas exchange after birth precarious.
When these categories of disease were first devised, severe fetal disease predicted severe neonatal disease. However, as fetal and neonatal management has been refined, the predictions must be updated, and the proportion of mildy and moderately affected neonates has risen. For example, some mildly anemic but not hydropic neonates who met previous criteria for exchange transfusion are now managed effectively with intensive phototherapy and would therefore be regarded as having mild rather than moderate disease. Similarly, a fetus with early development of hydrops (severe HDFN) may respond well to one or more intrauterine transfusions and therefore be born with mild neonatal disease and yet still have severe late anemia that is as much a consequence of hematopoietic suppression caused by effective intrauterine treatment as of hemolysis caused by the underlying disease. A suggested update to the working definitions that separates the criteria into epochs and includes a summary of implications for practice is given in Table 3-4 .
| Severity | Mild | Moderate | Severe |
|---|---|---|---|
| In Fetus | |||
| Historical proportion of Rh HDFN before intrauterine therapy was available | ~50% | 25%-30% | 20%-25% |
| Likely history in previous siblings | No exchange transfusion | Exchange transfusion unless in utero treatment | Death unless in utero treatment |
| Likely maternal anti-D titer | <1 : 64 | >1 : 64 | >1 : 64 |
| Ultrasound abnormalities | None | Altered patterns of flow in fetal vessels | Signs of severe anemia (see Table 3-5 ), which may lead to hydrops, portal hypertension, ascites and other effusions, polyhydramnios |
| In Neonate | |||
| Clinical condition at birth | Normal | Mildly or moderately ill | Hydrops |
| Hemoglobin at birth | No anemia | Mild, or moderate anemia; Hg >0.6 MoM for gestation | Severe, symptomatic anemia Hg <0.6 MoM |
| Hemoglobin nadir | Transfusion not needed | At risk for symptomic anemia | Risk of death unless transfusion |
| Bilirubin cord | <3.5 mg/dL (≈70 µmol/L) | >3.5 mg/dL (≈70 µmol/L) | >3.5 mg/dL (≈70 µmol/L) |
| Bilirubin peak | Term: <20 mg/dL (≈380 µmol/L) Premature: lower levels depending on prematurity | Term: >20 mg/dL (≈380 µmol/L) Premature: lower levels depending on prematurity | Rate of RBC destruction depends on (low) hemoglobin B; peak bilirubin is variable, toxicity increased |
| Phototherapy | If bilirubin increases faster than 0.4-0.5 mg/dL/hr; lower threshold for premature infants | If bilirubin increases faster than 0.4-0.5 mg/dL/hr; lower threshold for premature infants | Immediate, intensive, pending measurement of bilirubin increase |
| “Early” exchange | No | If rate of increase of bilirubin >0.8-1 mg/dL/hr despite several hours of intensive phototherapy; lower threshold for premature infants | If rate of increase of bilirubin >0.8-1 mg/dL/hr despite several hours of intensive phototherapy; lower threshold for premature infants |
| “Late” exchange | If bilirubin > exchange level | If bilirubin ≥ exchange level | If bilirubin ≥ exchange level |
| Heme oxygenase inhibition | Unnecessary; number needed to treat to avoid exchange will be high | In context of well-designed clinical trials | In clinical trial to avert late exchange; unlikely to allow avoidance of early exchange |
| Intravenous immunoglobulin | Unnecessary; number needed to treat to avoid exchange will be high | Evidence from high-quality trials to date does not support; therefore only in context of further well-designed randomized trials | Evidence from high-quality trials to date does not support; therefore only in context of further well-designed randomized trials |
| Phenobarbital | No | Neonatal—no clear benefit if optimal phototherapy; may be role for antenatal treatment in context of well-designed clinical trials | Neonatal—no clear benefit if optimal phototherapy; may be role for antenatal treatment in context of well-designed clinical trials |
| Erythropoietin (or analogue) for late anemia | No | In context of well-designed clinical trials | In context of well-designed clinical trials |
Pathogenesis of Hydrops
Hydrops in HDFN was originally attributed to fetal heart failure, but other factors probably play a role. With severe hemolysis and progressively greater extramedullary erythropoiesis, the hepatic architecture and circulation are distorted by the islets of erythropoiesis. It has been suggested that this results in portal and umbilical venous obstruction and portal hypertension. Impairment of umbilical venous return would exacerbate the fetal tissue hypoxia caused by anemia and explain the placental edema that is usually seen in severe hydrops. The placental edema, by increasing the placental barrier to gas exchange, could then impair oxygen delivery even more. The theory that liver distortion and dysfunction are at least as important as heart failure in the pathogenesis of hydrops fetalis could explain why hydrops is not always accompanied by hypervolemia or even severe anemia. Support for the suggestion that portal hypertension is present is provided by ultrasound assessment of waveforms in the portal venous system.
Hypoalbuminemia is a common feature, but the mechanism has not been explained. It could be caused by a greater volume of distribution for albumin or reversion to α-fetoprotein expression rather than failure of hepatic synthesis.
The mechanisms that lead to hydrops, both in anemic fetuses and in nonanemic fetuses with “nonimmune” hydrops, remain poorly understood. Various inflammatory mediators could play a role but have not been examined. Genes that are regulated by hypoxia include those for vascular endothelial growth factor and other molecules involved in capillary morphogenesis. Depending on the pattern of expression, these molecules can markedly affect capillary permeability and could also play a role.
Factors Affecting the Severity of Hemolytic Disease of the Fetus and Newborn
If a sensitized mother is pregnant with an Rh-incompatible fetus, a number of factors influence the severity of disease in the fetus. These factors include placental transfer of antibody, antibody characteristics, antigen characteristics, state of maturation of the fetal spleen, and resilience of fetal erythropoiesis.
Transfer of Antibody to the Fetus
All four IgG subclasses are transported across the placenta, via FcRn receptor–mediated endocytosis and then exocytosis. The fetal-to-maternal ratio for total IgG is approximately 0.4 at 28 weeks’ gestation and increases to 1.4 at term, but fetal levels of IgG1 are close to maternal levels by 17 to 20 weeks’ gestation. The efficiency of placental transport of antibody may be an important factor in the severity of Rh HDFN, and reduced IgG transport can result in unexpectedly mild disease.
Mechanism of Red Blood Cell Hemolysis
When anti-RBC antibodies bind complement and are present in high concentrations (as with transfusion reactions to ABO-incompatible blood in adults), RBC intravascular destruction occurs and may cause a clinically severe reaction including hemoglobinemia, shock, and disseminated intravascular coagulopathy. Products of RBC destruction are cleared predominantly by the liver.
When antibodies do not fix complement, as is typical with anti-RhD in the fetus and neonate, the mechanisms of hemolysis are more subtle but, in the end, equally destructive. Three Fc receptor–mediated pathways have been implicated in the destruction of opsonized RBCs: direct phagocytosis, phagocytic damage leading to later lysis, and killer cell–mediated damage. When anti-RhD becomes concentrated on the membrane of RhD-positive RBCs, the RBCs adhere to macrophages and form rosettes. This occurs particularly in the red pulp of the spleen, where RBCs and macrophages come into close apposition. The phagocytic cell can then destroy the RBC or damage and deplete the membrane, causing loss of RBC deformability. If the RBC escapes being consumed by the macrophage, its membrane is damaged, and its osmotic fragility and likelihood of lysis are greater.
Antibody-dependent cellular cytotoxicity (ADCC) probably also accounts for the destruction of some RBCs. Large granular “K cells” (natural killer lymphocytes) or myeloid cells (polymorphonuclear leukocytes and monocytes) bind antibody-labeled cells and release lysosomal enzymes and perforin, causing lysis.
Hemolysis also requires a competent system of phagocytic cells in the fetus. The clinical manifestations of the disease suggest that their maturation is sufficient as early as the middle of the second trimester.
Antibody Specificity and Severity of Hemolytic Disease
Antigen characteristics, including details of antibody distribution, density, and structure, all affect risk for HDFN; these have been reviewed by Hadley. Antibodies with the potential to cause HDFN generally recognize antigens restricted to erythroid cells (e.g., Rh antigens) rather than those with a wider tissue distribution (e.g., A and B antigens, found on many cell types). The antigens must be expressed in the fetus and neonate. Thus antibodies to Kell and Rh antigens, which are expressed early in fetal life, cause severe HDFN, whereas antibodies to Lewis antigens, which are not synthesized in erythrocyte progenitors, never cause HDFN. The circulating glycosphingolipids carrying the antigenic Le a or Le b epitopes generally only adsorb to the erythrocyte membrane during postnatal life. Lewis antibodies are also usually IgM. Lewis is the only human blood group system that has never been reported to cause HDFN of any severity.
Antigen density is also an important factor. Within the Rh system, even in the presence of high maternal anti-RhD titers, fetuses with weak RhD phenotypes are unlikely to be severely affected. As might be expected, homozygotes for RHD appear to have higher RhD antigen density than heterozygotes, although this is only potentially relevant in unusual situations such as ovum donation, because otherwise RhD positive infants of negative mothers are obligate heterozygotes. RHCE genotype also affects RhD antigen density, and in particular there seems to be higher expression of D where CE is deleted or underexpressed. The density of several other antigens, including A and B and the Lutheran antigens, is developmentally regulated, and antigen density on immature RBCs limits the severity of hemolysis. Antigen structure may also play a role, with some antigens being more capable of promoting recognition of RBCs by phagocytic cells.
Antibody characteristics also play a role in determining the severity of HDFN. IgG subclasses bind different classes of Fc receptors, although it has not yet been established conclusively which receptors are involved in RBC destruction in vivo. Fc receptor polymorphisms that bind IgG more avidly (and may play a role in ABO HDFN ) may yet be found to account for some of the heterogeneity of Rh HDFN.
Whereas anti-A and anti-B antibodies are predominantly IgG2, IgG1 and IgG3 anti-D antibodies tend to predominate in Rh-sensitized women. The different IgG subclasses are often present in combination.
The capacity of RBC-bound IgG3 antibodies to bind to Fc receptors of monocytes is greater than that of IgG1 antibodies. Threshold levels of antibody binding for rosette formation in vitro are approximately 1000 IgG3 or 2000 IgG1 molecules per RBC. Both can promote phagocytosis, but their relative potencies depend on circumstances. IgG3 usually causes more efficient lysis by monocytes than does IgG1. Clearance of Rh-positive RBCs is caused by fewer molecules of IgG3 anti-D than of IgG1 anti-D. However, there is some evidence that IgG1 is more important in the pathogenesis of severe HDFN.
Human leukocyte antigen (HLA) antibodies have been found in the sera of some women who have infants with mild HDN despite high antibody levels. The HLA antibodies may block antibody-antigen interactions on the macrophage membrane.
Antibody Detection
ABO and RhD typing plus antibody screening is recommended at the first prenatal visit in all pregnancies, including visits for elective abortion. A history of prior pregnancies and blood transfusions should also be taken. If the blood group is D-negative or weak D, another antibody screening is recommended at 24 to 28 weeks’ gestation, before administration of anti-D immunoprophylaxis. If an alloantibody is detected at a low titer, screening is usually repeated at regular intervals to detect any increase. There are a variety of methods for assessing maternal antibodies, some that are better suited for screening methods and some that are more useful for predicting severity of disease.
Manual Methods
Coombs serum is an anti–human globulin produced by immunizing another species. Rh-positive RBCs are incubated with the serum sample that is being tested for the presence of anti-RhD. If anti-RhD is present in the sample, it adheres to the RhD-positive RBC membrane. The RBCs are then washed to remove nonadherent human protein and suspended in Coombs serum. If the RBCs are coated with anti-RhD, they are agglutinated by the antibodies to human immunoglobulin (positive test result). The reciprocal of the highest dilution of maternal serum that produces agglutination is the indirect antiglobulin titer.
Incubation of RBCs with enzymes such as papain, trypsin, and bromelin reduces the electrical potential of the RBC membranes, allowing agglutination of Rh-positive cells by IgG anti-D even in saline. Enzyme screening methods are the most sensitive manual techniques available for detecting Rh sensitization, but they detect many antibodies that have no clinical significance.
These manual methods have the advantages of sensitivity and flexibility, but the correlation between antibody titer and disease severity is poor.
Automated Analysis
Autoanalyzer methods have been developed and refined for more efficient and reproducible detection and quantitation of Rh and other antibodies. Autoanalyzer techniques are very sensitive, and the finding of an Rh antibody detected only by an autoanalyzer method does not necessarily indicate the presence of HDFN or even true sensitization, especially if the titer is very low and stable. However, rising titers indicate the need for urgent assessment of the fetus.
Prediction of Fetal Hemolytic Disease
Once sensitization is detected, subsequent investigative measures are required. To refine the estimation of risk, one or more of the following may need to be considered.
Fetal blood type is determined by the following:
- •
Paternal blood typing
- •
Fetal blood typing
Severity of disease is predicted by the following:
- •
History indicating the severity of HDFN in previous infants
- •
Maternal antibody quantitation
- •
Fetal ultrasound
- •
Amniotic fluid spectrophotometry
- •
Percutaneous fetal blood sampling
Determination of Fetal Blood Type
Paternal Blood Typing
In some instances paternal blood typing can indicate that the baby has no risk for HDFN. This situation occurs if the father is Rh negative and the mother was sensitized not by his offspring but by those of a previous partner or by transfusion. If the father is predicted to be Rh negative or heterozygous, the fetus may be completely unaffected regardless of the maternal antibody titer. In predominantly white populations, approximately 56% of Rh-positive individuals are heterozygous. Because the RHD and RHCE genes are closely linked, paternal RHD zygosity can be estimated from his Rh c/C and e/E type and his race, or more accurately using molecular techniques.
Fetal Blood Typing
Fetal Rh typing by genotyping, using DNA amplification or microarray techniques, is becoming increasingly feasible and widespread and is supplanting the need for antigen typing of fetal blood. Although the need for it can be obviated if paternal homozygosity is certain, it can be an especially important technique when paternity is uncertain or the father is heterozygous.
Molecular methods are now used routinely and with high accuracy in many locations to detect fetal RHD , RHCE , KEL , DARC (Duffy), and SLC14A1 (Kidd) genes in free fetal DNA in maternal blood, obviating the need for chorionic villus biopsy or amniocentesis to obtain fetal DNA. Cell-free fetal DNA, presumed to be from apoptotic syncytiotrophoblast, is now most commonly used for fetal genotyping because unlike some fetal cells found in the maternal circulation, it is cleared rapidly after delivery and therefore reflects only the current pregnancy. Fetal genotyping has the potential to reduce or predict the need for assessment throughout the pregnancy, including the need for invasive tests, and to avert the need for RhD immunoglobulinduring some pregnancies. Single-cell preimplantation diagnosis has also been described.
The genetic diversity of the Rh blood group system outlined in an earlier section of this chapter provides an important reminder to use caution in choosing primers that minimize false-positive and false-negative test results and to keep in mind the fact that genotype does not always obviously predict phenotype. The prevalence of RHD-RHCE hybrids and partially intact but functionally inert RH genes in certain populations can markedly reduce the validity of tests designed for application in predominantly white populations. The use of appropriate controls; amplification of at least two diagnostic sites, preferably checking the correlation between parents’ genotype and phenotype in parallel; and performance of the tests with a good understanding of the typical genotypes in the populations for which they were developed and to which they are being applied should minimize errors. The need to have a good positive control for fetal DNA to identify false-negative results is critical. The SRY sequence can be used if the fetus is male, but more complicated comparison of multiple fetal and parental polymorphisms in other genes may be necessary if the fetus is female. Differential methylation of the maspin gene in fetal and maternal tissues recommends it as a universal positive control.
RHD genotyping of amniotic and chorionic cells agrees with serologic and tissue typing, allowing CVS or amniocentesis to be used for typing of the fetus. However, the need to use these procedures should diminish as procedures using fetal DNA from maternal blood become more widely available. Some advantages of diagnosis using maternal blood samples are the ability to type early in pregnancy and the fact that sample acquisition neither risks harming the fetus nor increases maternal exposure to fetal antigens.
Prediction of Severity of Disease
The relative roles of methods for assessing the severity of hemolytic disease in the fetus have changed in recent years.
Pregnancy History
Although it is usually true that the severity of HDFN remains the same or increases during subsequent affected pregnancies, the disease sometimes becomes less severe. If a previous baby was hydropic, a subsequent affected fetus has a 90%, but not a 100%, chance of developing hydrops. If hydrops is going to develop, usually it does so at the same gestation or earlier, but occasionally it develops later. In a first RhD-sensitized pregnancy, with no prior history of HDFN, there is an 8% to 10% probability that hydrops will develop. If a mother has had a previous fetus with hydrops and the father is heterozygous for the offending antigen, the fetus may be negative and completely unaffected or positive and very severely affected, a dilemma that requires resolution, usually by fetal typing. Thus there are numerous situations in which the history of previous pregnancies is unreliable, although it is nevertheless worth noting.
Maternal Antibody Titers
Although antibody titrations carried out in the same laboratory by the same experienced personnel using the same methods and test cells are reproducible and do give the physician some indication of risk, their predictive value is inadequate to guide invasive fetal treatment. Use of automated analyzers provides somewhat better prediction of the severity of HDFN than manual methods do, but the two approaches have not been compared rigorously. Hadley has noted that whereas many laboratories can and should undertake screening for alloantibodies, antibody quantitation is best limited to regional centers with proven reliability. Ideally, these laboratories will also have strategies for auditing the outcomes of pregnancies in which antibodies have been detected. This can lead over time to the recognition of a critical titer, below which no cases of severe disease have occurred; this allows targeting of more extensive and invasive tests to those with titers above the threshold. However, the follow-up needed is laborious, prone to ascertainment bias, and possibly a violation of patient privacy. Generally, if the obstetric history is good, low titers (≤1 : 16 by manual methods or <5 IU/mL [1 µg/mL] by autoanalyzer) that increase slowly or not at all are reassuring and indicate continued surveillance using antibody titers and ultrasound.
Although rising titers suggest increasing risk to the fetus, it has not been possible, even with autoanalyzer methods, to establish a titer above which the fetus must be sensitized, and sometimes antibody levels increase significantly (but presumably nonspecifically) despite the presence of a compatible and therefore unaffected fetus. Nevertheless, in the presence of high or rising titers, fetal assessment becomes urgent.
Amniotic Fluid Spectrophotometry
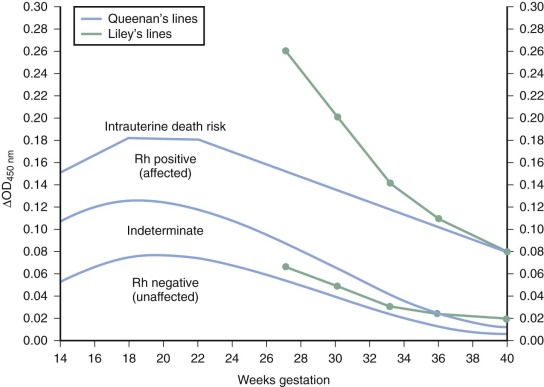
Bevis showed that bilirubin could be found in the amniotic fluid of infants with HDFN, and in 1961 Liley plotted fetal outcomes by gestation and results of amniotic fluid spectrophotometry, thus devising a prenatal test of severity of HDFN that has only recently been surpassed as the standard method of assessing fetuses with hemolytic disease. Liley used the ΔOD 450 reading (the deviation from linearity at 450 nm caused by the absorption spectrum of bilirubin) as a measurement of the amniotic fluid bilirubin level. Three zones of predicted severity of disease were defined ( Fig. 3-5 ). Readings in zone I indicated either no disease or no anemia at the time of testing but reflected a 10% chance that exchange transfusion would be needed. Readings in zone II indicated moderate disease that becomes more severe as readings approach the zone III boundary. Readings falling into very high zone II or zone III indicated severe disease: hydrops was present or would develop within 7 to 10 days. When serial ΔOD 450 measurements are taken, the overall accuracy of prediction of hemolytic disease with the amniotic fluid technique is 95%. Accuracy is higher in the third trimester than in the second trimester. Because the graphs were developed before intervention at earlier than 27 weeks’ gestation was contemplated, zone boundaries in the second trimester were not defined. The Liley zone boundaries were subsequently modified by inclining them downward before 24 weeks’ gestation because of the observation that ΔOD 450 readings peak at 23 to 24 weeks’ gestation in pregnancies unaffected by HDFN. The division lines for Queenan’s chart are lower at all gestations than those of Liley’s system. Although it was suggested that Queenan’s modification could overestimate severity, leading to too many interventions, others concluded that a more conservative approach was appropriate as safer and more effective intervention became available. Monitoring by ultrasound and selective direct fetal blood sampling has markedly reduced the need for assessment by amniocentesis. Amniocentesis may still sometimes be useful as an adjunct to ultrasound to assess severity. Increases in antibody titer occur after some amniocenteses, although the risk is reported to be lower than that after umbilical blood sampling.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


