Fig. 2.1
Timeline of BCR-ABL findings and development of imatinib
2.2 Molecular Biology of CML
2.2.1 Ph Chromosome
The Ph chromosome results from a reciprocal translocation, which involves the BCR gene on chromosome 22 at band q11 and the ABL proto-oncogene on chromosome 9 at band q34, t(9; 22)(q34; q11) [4] (Fig. 2.2a). Depending on the breakpoints in the BCR gene, there are three different forms of the BCR-ABL oncogene: p185/p190 (m-bcr breakpoint), p210 (M-bcr breakpoint), and p230 (μ-bcr breakpoint) (Fig. 2.2b, c). p210 BCR-ABL is observed in 90% of CML, p185/190 BCR-ABL in 20–30% of ALL, and p230 BCR-ABL in a subset of patients with chronic neutrophilic leukemia (CNL) patients [16].
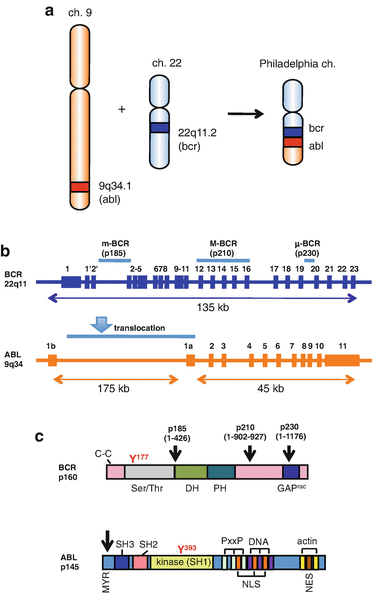
Fig. 2.2
Philadelphia chromosome (a) and localization of the breakpoints in the ABL and BCR genes (b) and proteins (c). a The fusion BCR-ABL gene is formed within Ph chromosome as a result of a reciprocal translocation between chromosomes 22 and 9. b BCR has three breakpoint cluster regions indicated as m-BCR, M-BCR, and μ-BCR, for p185/190, p210, and p230 BCR-ABL genes, respectively. The ABL gene has alternately spliced first exons Ib and Ia. An arrowhead indicates the region of the translocation. c BCR consists of the coiled-coil (C–C) domain and a region exhibiting serine/threonine kinase activity in the N-terminus region. Y177 is the autophosphorylation site crucial for binding to growth factor receptor-bound protein 2 (Grb2). The center region contains a dbl-like domain (DH) and pleckstrin homology (PH) domains. Toward the C-terminus is a domain with an activating function for Rac-GTPase (Rac-GAP). ABL consists of a myristoylation (MYR) site and SH3 and SH2 domains in the N-terminus region. Y393 is the major site of autophosphorylation within the kinase (SH1) domain. The middle region consists of proline-rich regions (PxxP), nuclear localization signal (NLS), nuclear export signal (NES), and DNA-binding region (DNA). The C-terminus contains G- and F-actin-binding domain (actin). Arrowheads indicate the position of the breakpoints in the p185/190, 210, and 230 BCR-ABL proteins, respectively
2.2.2 Structure and Intramolecular Interaction of BCR-ABL
The structure of p210 BCR-ABL is shown in Fig. 2.3a. The tyrosine kinase c-ABL is negatively regulated by interactions between its SH3 and SH2 domains and the distal surface of the kinase domain. The myristoyl group of c-ABL is inserted into the hydrophobic pocket of the kinase domain to dock of the SH2 and SH3 domains onto the kinase domain [17]. The interface between the SH3 domain and the linker segment connecting the SH2 and the kinase domains also provides negative regulation [18]. In contrast, BCR-ABL exists primarily as a dimmer or tetramer to facilitate trans-autophosphorylation. The N-terminal coiled-coil oligomerization domain in the BCR sequence plays an essential role in the activation of BCR-ABL, which is similar to ligand-induced dimerization and subsequent activation of receptor tyrosine kinases [19, 20]. In an activated state of BCR-ABL, the inhibition through the SH3 domain and the linker region is released, and the SH2 domain not only binds the N-lobe via tight interactions between I145 and T272/Y312 but also the SH2-binding domain in the BCR sequence as well as SH2 ligands [21] (Fig. 2.3b).
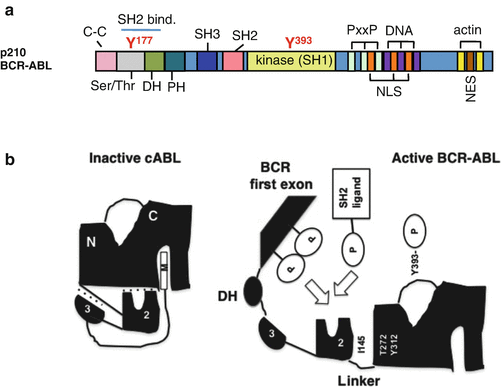
Fig. 2.3
Structure and intramolecular interaction of BCR-ABL. a The p210 BCR-ABL protein contains the coiled-coil (C–C) domain, Ser/Thr kinase domain, dbl-like domain (DH), and pleckstrin homology (PH) domains from BCR, as well as the SH domains, proline-rich (PxxP), nuclear localization signal (NLS), nuclear export signal (NES), DNA-binding (DNA), and actin-binding domains (actin) from ABL. The tyrosine residues in the Ser/Thr (Y177) and SH1 kinase (Y393) domains have been highlighted. b Intramolecular inhibitory folding through the interactions between domains is shown with dotted lines, including binding between (1) the SH3 domain and the SH2 linker, (2) the SH2 and the tyrosine kinase domains, and (3) the myristoyl group in N-terminal ABL and the tyrosine kinase domain. The inhibitory folding in the inactive state in c-ABL is released in BCR-ABL, allowing autophosphorylation in the activation loop at Y393 and the interaction between (1) the SH2 domain and the N-lobe and (2) the SH2 ligands and the SH2 domain (see details in the text; Maru [22])
2.2.3 Signaling Pathways Downstream of BCR-ABL
The deregulated BCR-ABL activates a large number of signal transduction pathways including Ras, signal transducer and activator of transcription-5 (STAT5), phosphatidylinositol 3-kinase (PI3), Myc, RhoA-Rac, and cyclin D1, which lead to uncontrolled proliferation and differentiation, resistance to apoptosis, cell migration, and unique metabolism and defect in DNA repair in progenitor cells [22] (Fig. 2.4).
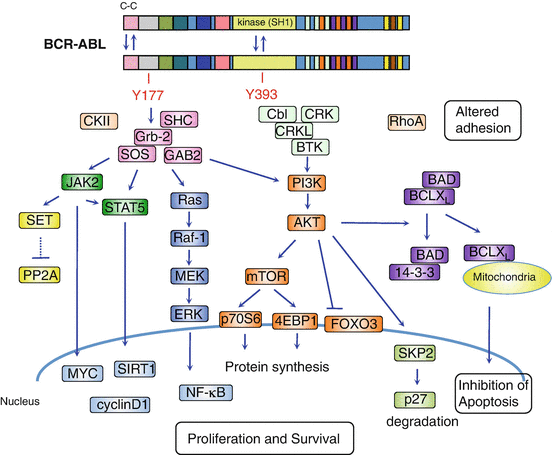
Fig. 2.4
Schematic representation of the downstream molecular pathways of BCR-ABL. Dimerization of BCR-ABL via the N-terminal coiled-coil domain activates BCR-ABL kinase leading to autophosphorylation of Y393 in ABL followed by Y177 in BCR. The phosphorylation of Y177 recruits Grb2, SOS (a guanine nucleotide exchanger of RAS), and Grb2-associated-binding protein 2 (GAB2) complex. This complex activates Ras/MAPK/ERK, phosphatidylinositol 3-kinase (PI3K)/AKT, and JAK/STAT signaling pathways. The activated AKT induces mTOR activation, suppression of forkhead O (FOXO) transcription factor, and proteosomal degradation of p27. PI3K-AKT also promotes phosphorylation of the proapoptotic protein Bad, leading to its interaction with chaperone protein 14-3-3, which in turn results in the restriction of Bad to the cytoplasm. This attenuates Bad opposing BCL2 and BCL-XL inhibition of apoptosis in the mitochondria. Both, through activation of JAK2, directly and indirectly, activate STAT5. JAK2 also activates the PP2A inhibitor, SET, leading to PP2A inhibition. These downstream signaling pathways of BCR-ABL promote proliferation and survival and inhibit apoptosis
2.3 Mechanism of BCR-ABL Inhibition by Imatinib
Imatinib acts as an ATP-competitive tyrosine kinase inhibitor (TKI) via interaction with the ABL kinase domain. Binding of imatinib prevents BCR-ABL’s ability to transfer a phosphate group onto tyrosine on the substrate and its subsequent activation, which blocks the transmission of proliferative signals to the nucleus, thus inducing apoptosis in the leukemic cell [23] (Fig. 2.5).
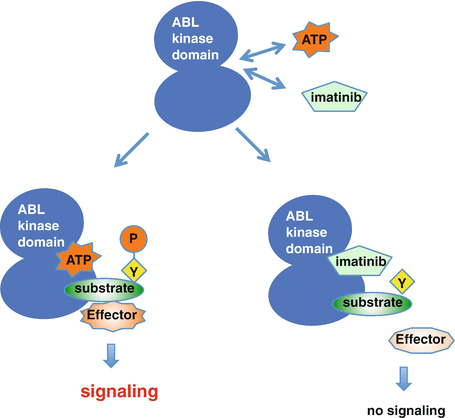
Fig. 2.5
Imatinib acts as an ATP-competitive inhibitor. The deregulated tyrosine kinase, BCR-ABL, functions by transferring phosphate from ATP to tyrosine residues on the substrates, which promotes downstream signaling pathways that lead to the stimulation of proliferation and inhibition of apoptosis. Imatinib competes with ATP at the BCR-ABL kinase domain and specifically inhibits the downstream signaling cascade
2.3.1 Imatinib: Chemical Structure
Imatinib is a 4-[(4-methyl-1-piperazinyl)methyl]-n-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]-benzamide monomethanesulfonate. N-methylpiperazine, as a polar side chain, is associated with good water solubility and oral bioavailability. The introduction of a flag-methyl group at the 6-position of the anilino phenyl ring increases the selectivity for ABL kinase. Cellular activity is enhanced by the addition of a 3′-pyridyl group at the 3′-position of the pyrimidine [15, 23] (Fig. 2.6).
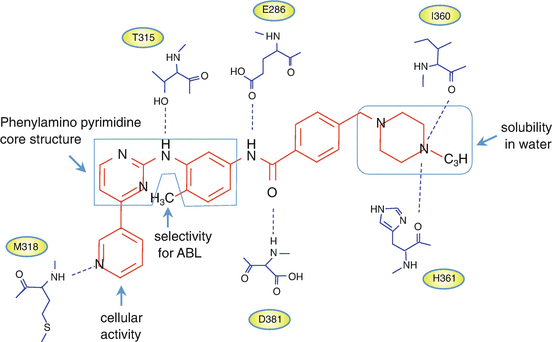
Fig. 2.6
Imatinib and ABL kinase domain interactions. Chemical structures of imatinib and schematic diagram showing the six hydrogen bonds between imatinib and the BCR-ABL kinase domain (see details in the text)
2.3.2 Imatinib and ABL Kinase Domain Interactions
The catalytic domain of protein kinase is well-conserved and consists of the N-lobe and the C-lobe. Imatinib functions through competitive inhibition of ATP at the ATP-binding site of the BCR-ABL kinase domain. The Asp-Phe-Gly (DFG) motif in the activation loop of the N-lobe, adjacent to the ATP-binding site, is very important for the kinase’s inhibitors. The αC-helix in the C-lobe contributes to an on-off switch related to the conformational status of “ready-to-phosphorylate substrates.” Type I and II inhibitors are the predominant groups of targeted protein kinase inhibitors [24]. The type I inhibitors recognize the active conformation of the kinase, while imatinib, which belongs to the type II protein kinase inhibitors, recognizes the inactive conformation of the enzyme [25]. The binding to a specific “DFG-out” conformation of BCR-ABL contributes to the selectivity of type II inhibitors. Imatinib interacts with 21 amino acid residues in the ABL kinase domain, forming six hydrogen bonds (Fig. 2.6). The aromatic ring of the inhibitor also interacts with the protein through van der Waals interactions [26].
2.3.3 Imatinib: Pharmacodynamic Properties
In vitro kinase assay showed that imatinib potently inhibits ABL kinase and its active derivatives, BCR-ABL and v-ABL with IC50 values in the range of 0.025 μM for autophosphorylation. Imatinib also inhibits the platelet-derived growth factor receptor (PDGFR) and Steel factor receptor (c-KIT) kinases at a similar concentration. On the contrary, a large number of other kinases including Src family kinases such as c-Src, c-Lyn, and c-Lck and the epidermal growth factor receptor (EGFR) exhibited at least 100-fold higher IC50 values [13, 23] (Table 2.1). According to the substrate specificity, imatinib is not only approved for the treatment of Ph+ CML and ALL but also for the treatment of KIT-mutant-positive gastrointestinal stromal tumor (GIST) and myelodysplastic diseases with PDGFR gene rearrangements and hypereosinophilic syndrome with Fip1-like-1 (FIP1L1)-PDGFRα fusion proteins [27].
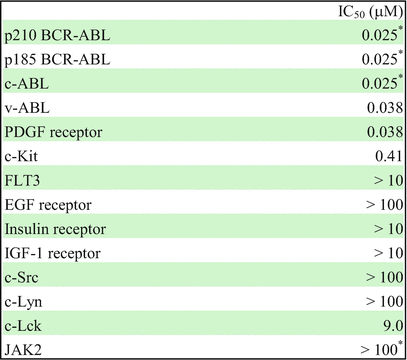
Table 2.1
Selectivity profile of imatinib
2.3.4 Effects of Imatinib on CML Mouse
Imatinib inhibits tumor growth in mice injected with BCR-ABL-transformed 32D cells and in nude mice injected with human BCR-ABL-positive KU812 cells [13, 28]. Imatinib significantly prolonged the survival of the murine bone marrow transduction-transplantation model of CML. Although all the animals exhibited prolonged survival after imatinib treatment, approximately 20% died of leukemia [29].
2.4 Imatinib Resistance and Targeting Strategies to Overcome the Resistance
Although 80% of patients quickly achieved a complete cytogenetic response, a small proportion of patients in the chronic phase (CP) and a substantial proportion in the accelerated phase (AP) and the blast phase (BP) failed to respond to imatinib. The precise mechanism of disease progression is still uncertain, but the additional genetic changes involving activation of the oncogenic factor and/or inactivation of the tumor suppressor appear to cause more advanced CML-AP or CML-BP [33] (Fig. 2.7). As CML stem cells are not addicted to BCR-ABL, imatinib failed to kill BCR-ABL-expressing CML stem cells [34, 35]. Several mechanisms of imatinib resistance have been reported, including BCR-ABL-dependent mechanisms (i.e., mutations in specific amino acids within ABL kinase or overexpression of the BCR-ABL protein) and BCR-ABL-independent mechanisms (i.e., drug import or export, additional genetic abnormalities, and the activation of alternative signaling pathways, such as Ras/Raf/MEK kinase, STAT or Src family kinases) [36, 37]. To overcome imatinib/TKIs resistance, several strategies are being investigated (Fig. 2.8, Table 2.2).
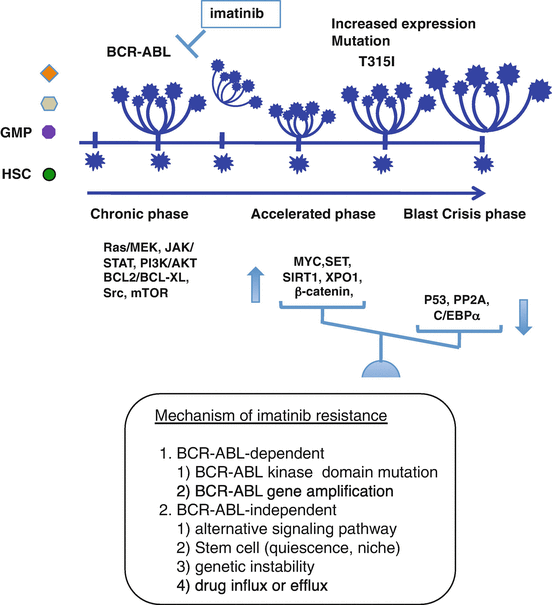
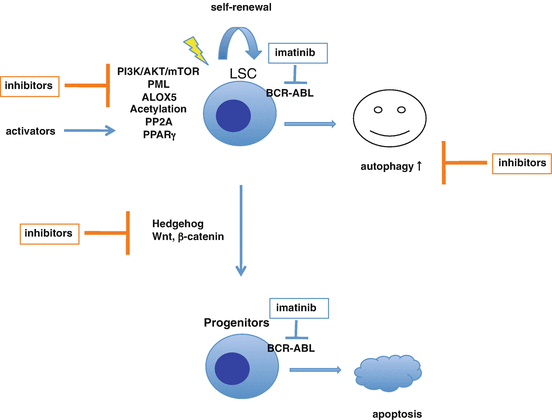
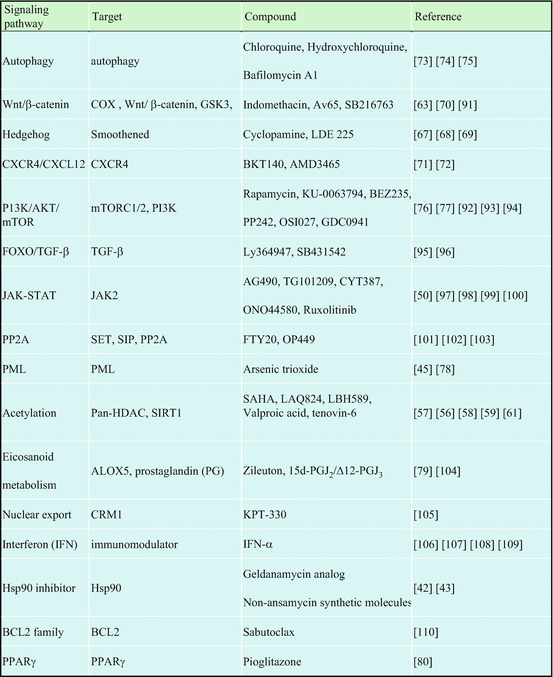
Fig. 2.7
Mechanism of imatinib resistance. Imatinib fails to kill BCR-ABL-expressing CML stem cells. During treatment with imatinib, accumulation of genetic changes involving activation of the oncogenic factor and/or inactivation of the tumor suppressor appears to induce more advanced CML-AP or CML-BP (Maru [22])
Fig. 2.8
Pharmacological targeting CML stem cells to overcome imatinib resistance. Imatinib effectively inhibits BCR-ABL kinase activity leading to apoptosis in the more mature progenitor cells. In contrast, the quiescent CML stem cells are less sensitive to imatinib, and furthermore imatinib-induced autophagy eliminates defective mitochondria, a source of reactive oxygen species, to promote cell survival. Autophagy inhibitors are under investigation alone and in combination with imatinib/TKI. Several inhibitors and activators targeting CML stem cells and bone marrow niche are also being investigated
Table 2.2
Target signaling pathways and compounds to overcome imatinib resistance
2.4.1 BCR-ABL-Dependent Imatinib Resistance
2.4.1.1 BCR-ABL Kinase Domain Mutations
The presence or acquisition of mutations in the BCR-ABL kinase domain has been shown to confer variable degrees of resistance to imatinib [38]. The mutations responsible for imatinib resistance mainly occur at seven sites: G250, Y253, E255 (in the phosphate-binding region or P-loop), T315I (in the imatinib-binding site), M351, F359 (near the catalytic domain), and H396 (in the activation loop) [39]. The substitution of the “gatekeeper” residue T315 with the bulkier and more hydrophobic isoleucine results in the loss of an important hydrogen bond between imatinib and the kinase domain. Second-generation TKIs, dasatinib and nilotinib, are more effective than imatinib in preventing the progression to accelerated phase and blast crisis. These drugs are highly potent against imatinib-resistant ABL kinase mutations, except T315I mutation. The third-generation TKI, ponatinib, can inhibit BCR-ABL with a T315I mutation [40].
2.4.1.2 Gene Amplification and Upregulation of BCR-ABL Protein
Gene amplification results in the overexpression of the BCR-ABL protein. Moreover, the binding of imatinib to the ABL kinase domain appears to stabilize the BCR-ABL protein [41]. Newly synthesized BCR-ABL proteins are initially stabilized by Hsc70 and then passed on to Hsp90 and cochaperone Cdc37 to achieve maturation or kinase activation. Hsp90 inhibitors can ubiquitinate and degrade BCR-ABL proteins with or without mutations (e.g., T315I and E255K) via an E3 ligase CHIP and a c-Cbl-mediated proteasome machinery [41, 42]. The second generation of Hsp90 inhibitors has entered clinical trials [43]. Protein phosphatase 2A (PP2A) agonists can inhibit BCR-ABL kinase, also leading to degradation of the BCR-ABL protein [44]. Arsenic trioxide (As2O3) downregulates BCR-ABL protein via an autophagic or a proteasomal degradation mechanism [45].
2.4.2 BCR-ABL-Independent Imatinib Resistance
2.4.2.1 Alternative Signaling Pathways
The ABL kinase domain-independent scaffolding function of BCR-ABL appears to activate pro-survival signaling pathways, leading to imatinib resistance. The Src family kinase, Hck, is activated by BCR-ABL in a kinase-independent manner, and the activated Hck can phosphorylate Y177 in BCR, leading to an activation of the STAT5 signaling pathway [46, 47]. Y177 phosphorylation is also induced by the reciprocal activation between JAK2 and BCR-ABL, which results in BCR-ABL protein stability and activation of STAT5, Ras, and Lyn-mediated stimulation of the PI3K-AKT pathway [48]. Highly activated Lyn has been observed in TKI-resistant patients [49]. Inhibition of both BCR-ABL and the Src family kinases by the second- and third-generation TKIs is expected to overcome the Src family kinase-dependent imatinib resistance. A non-ATP-competitive dual inhibitor for JAK2 and BCR-ABL is effective in T315I BCR-ABL [50].
2.4.2.2 Genetic Instability
BCR-ABL appears to induce genomic instability in CML-CP stem cells during imatinib treatment, which may result from an aberrant cellular response to enhanced DNA damage caused by higher levels of reactive oxygen species (ROS) [51]. The inefficient repair generates not only TKI-resistant point mutations in BCR-ABL kinase but also point mutations in other genes (e.g., p53 and Ras). BCR-ABL-induced deregulated mechanisms of DNA repair appear to involve downregulation of the catalytic subunit of DNA-PK, disruption of the Fanconi anemia/BRCA DNA repair pathway, activation of Rad51 and the Werner syndrome helicase (WRN), and inhibition of uracil DNA glycosylase UNG [52–55]. In addition, the genetic mutations may tend to occur through BCR-ABL-induced alterations of epigenetic regulators involving histone acetyltransferases and deacetylases (HDAC). HDAC inhibitors enhance imatinib-induced apoptosis of CML cells [56–60]. Inhibition of SIRT1, an NAD-dependent histone deacetylase, enhances p53 acetylation leading to its transcriptional activity, which promotes elimination of CML stem cells following imatinib treatment [61].
2.4.2.3 Stem Cells and Bone Marrow Niche
As mentioned above, imatinib is unable to eradicate quiescent CD34+ CML stem cells [34, 35]. In the hypoxic microenvironment of the hematopoietic stem cell (HSC) niche, HIF1-α signaling supports CML stem cells persistence independent of BCR-ABL kinase activity [62]. Wnt-β-catenin signaling leads to self-renewal in stem cells by activating the expression of Myc and cyclin D1 [63]. Aberrant activation of Wnt signaling endows granulocyte-macrophage progenitor cells (GMPs) with self-renewal capacity in CML-BC [64]. A close interplay between the Wnt-β-catenin pathway and N-cadherin in the bone marrow mesenchymal stromal cell also protects CML stem and progenitor cells during TKI treatment [65]. The Hedgehog pathway is a developmental signaling pathway that also plays a role in primitive and adult hematopoiesis [66]. BCR-ABL-induced upregulation of Smo activates the Hedgehog pathway in CML stem cells via the Gli family of transcriptional effectors. The inhibitors for the Wnt and the Hedgehog signaling pathways in combination with imatinib are expected to eradicate CML stem cells [63, 67–70].
The bone marrow microenvironment has been implicated in the protection of CML stem cells from imatinib-induced apoptosis. CXCL12, also called stromal cell-derived factor-1 (SDF-1), a chemokine produced by bone marrow stromal cells, and its receptor CXCR4 regulate HSC migration to and from the bone marrow. BCR-ABL downregulates CXCR4 expression, leading to a defective adhesion of CML cells to bone marrow stroma. Imatinib-induced upregulation of CXCR4 promotes migration of CML cells to bone marrow stroma, which results in the survival of quiescent CML progenitor cells [71]. CXCR4 inhibitor-induced disruption of stroma-mediated protection enhances imatinib sensitivity [72].
Autophagy appears to act as a double-edged sword for tumor cells. Autophagy selectively removes damaged organelles, especially dysfunctional mitochondria that are the source of reactive oxygen species. BCR-ABL-induced activation of the PI3K-AKT-mTOR signaling pathway inhibits autophagy, which results in high ROS levels that induce genome instability [73, 74]. On the contrary, treatment with imatinib induces autophagy that is associated with endoplasmic reticulum stress, which results in stem cell survival. The combination of TKI and an autophagy inhibitor resulted in a dramatic growth inhibition of CD34+ cells from CML patients [75]. The mTOR inhibitor, rapamycin, also decreased the viability of K562 cells and can block cell proliferation in imatinib-resistant CML patients [76, 77].
The promyelocytic leukemia protein (PML) tumor suppressor, which acts as a negative regulator of mTOR, plays a critical role in HSCs. As2O3 induces PML degradation and thereby inhibits maintenance of CML stem cells [78]. BCR-ABL induced upregulation of arachidonate 5-lipoxygenase (ALOX5), which is not inhibited by imatinib, suggesting that kinase independence is essential for the induction of CML in mice. The ALOX5 inhibitor-induced depletion of CML, but not normal, stem cells, and imatinib-induced elimination of differentiated leukemia cells could be a good combination [79]. Glitazone-induced activation of peroxisome proliferator-activated receptor-γ (PPARγ) has recently been shown to decrease STAT5 expression and that of its downstream key molecules such as HIF2α and CITED2 in quiescent stem cells. The combination of imatinib and pioglitazone can synergistically decrease CML stem cells [80].
2.4.2.4 Imatinib Influx or Efflux
Imatinib is a substrate of the influx transporter, organic cation transporter 1 (OCT1) and the efflux transporters, P-glycoprotein (P-gP, ABCB1, or MDR1), and breast cancer-related protein (BCRP or ABCG2) [81–83]. Increased efflux and decreased influx of imatinib may be associated with the imatinib resistance. Several studies have shown a clinical determinant of intracellular drug levels and resistance to imatinib [81]. An ABCB1 inhibitor, cyclosporine, resensitized imatinib-resistant K562 cells to imatinib in vitro [84]. Statins also inhibited ABCB1 and ABCG2 efflux pump activity, which resulted in potentiation of antileukemic activity of imatinib in primary CD34+ CML-CP and CML-BP cells [85].
2.5 Effects of Imatinib on Other Tyrosine Kinases
Imatinib has been shown to be able to inhibit other tyrosine kinases including c-ABL, PDGFR, and c-Kit. Thus, imatinib may be used to treat various other tumors caused by imatinib-specific abnormalities of PDGFR and c-Kit [86]. Imatinib also affects energy metabolism, bone metabolism, or endocrine pathways [87]. Imatinib appears to cause dysregulation of bone remodeling through attenuation of osteoclasts via inhibition of c-Kit, c-fms, and PDGFR and activation of osteoblast activity via inhibition of PDGFR [88]. c-ABL participates in the regulation of the endothelial barrier. Inhibition of c-ABL by imatinib causes a reduction of endothelial permeability in response to VEGF and the inflammatory mediators, thrombin, and histamine [89]. PDGF is involved in cardiovascular diseases, such as atherosclerosis, pulmonary arterial hypertension, angiogenesis, and diabetes. Inhibition of PDGFR by imatinib may improve the cardio-metabolic dysfunctions [90].
References
1.
Piller G. Historical review LEUKAEMIA – a brief historical review from ancient times to 1950. Br J Haematol. 2001;112:282–92.
2.
Nowell P, Hungerford D. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;132:1497.
3.
Fialkow PJ, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci U S A. 1967;58(4):1468–71.PubMedPubMedCentral
4.
Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature. 1973;243(5405):290–3. doi:10.1038/243290a0.PubMed
5.
Abelson HT, Rabstein LS. Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Res. 1970;30:2213–22.PubMed
6.
Bartram CR, de Klein A, Hagemeijer A, van Agthoven T, Geurts van Kessel A, Bootsma D, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277–80.PubMed
Related posts:
 Bcr-Abl Tyrosine Kinase Inhibitors (TKIs)
Inotuzumab Ozogamicin for Acute Lymphoblastic Leukemia: Clinical Pharmacology and Therapeutic Results
Therapies Targeting Leukemic Stem Cells
Bcr-Abl Tyrosine Kinase Inhibitors (TKIs)
Inotuzumab Ozogamicin for Acute Lymphoblastic Leukemia: Clinical Pharmacology and Therapeutic Results
Therapies Targeting Leukemic Stem Cells
 Nelarabine
Nelarabine
 Clofarabine: Structure, Mechanism of Action, and Clinical Pharmacology
Clofarabine: Structure, Mechanism of Action, and Clinical Pharmacology
 Imatinib: Clinical Pharmacology and Therapeutic Results
Imatinib: Clinical Pharmacology and Therapeutic Results
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


