Fig. 17.1
Sites of IDO/TDO expression and action in cancer. Expression of IDO1, IDO2, and TDO documented in various cells in the tumor microenvironment (including metastatic sites) and in tumor-draining lymph nodes (TDLN) is indicated, including in tumor, stromal, vascular, and immune cells. Both tryptophan deprivation and kynurenine production mediated by IDO1 and TDO have been implicated in inflammatory processes and immune escape (antigenic tolerance). Effects of IDO/TDO activity on the function of T cells and MDSC are shown. APC antigen-presenting cell (e.g., dendritic cell), MDSC myeloid-derived suppressor cell, TAM tumor-associated macrophage, TAN tumor-associated neutrophil, Teff T effector cell, Treg T regulatory cell
17.2 IDO1 in Immune Escape from T-Cell Immunity
The prevailing view among cancer biologists of the determinative importance of intrinsic tumor cell characteristics was encapsulated in a highly influential categorization of the hallmarks of cancer [27]. In this broad conceptualization, even metastasis and angiogenesis, the two recognized hallmarks with clear host dependence, were considered from a tumor-centric perspective, and no consideration was given to possibility that interactions with host immunity might also play an instrumental role in cancer outcomes. The case for including immune escape within the pantheon of critical hallmarks was first promulgated in a 2008 review on IDO1 [20] and eventually gained general acceptance as the hallmark designations were reassessed in light of recognition of the importance of host environmental factors such as immunity and inflammation [28, 29].
IDO1 induction in DC and macrophages promotes immune tolerance by suppressing effector T cells, converting naïve T cells to FoxP3+ Tregs, and elevating the suppressive activity of “natural” Tregs [30]. Extratumoral induction of IDO1 was reported initially in a subset of cancer patients and preclinical tumor graft models [15, 31]. In the mouse B16 melanoma model, IDO1 was not detectable directly in the tumors that formed but rather was elevated in TDLN where it was localized to a specific subset of DC characterized for T-cell suppressive activity [31]. Several different IDO1 inhibitory compounds have since been identified that can produce highly significant B16 tumor growth suppression that relies both on intact T-cell immunity and host IDO1 function [20, 22, 32], providing pharmacological support for an extratumoral role of IDO1 in limiting antitumor immunity. In like manner, the first genetic validation of IDO1’s involvement in driving autochthonous tumor development came from studies in classical two-stage models of skin carcinogenesis, where there was no evidence of IDO1 expression in the developing lesions: similar to the B16 model, IDO1 expression and activity were highly elevated in DC within the TDLN [24]. In this context, where tumor initiation and promotion are distinctly separable, IDO1 was found to be elevated in the tumor-promoting inflammatory environment, even in the absence of tumor initiation, clearly indicating that extratumoral IDO1 elevation is an early event that occurs before initiation in programming a pro-tumorigenic inflammatory microenvironment [33].
17.3 IDO1 in Inflammatory Programming: MDSC Development and Metastasis
Myeloid-derived suppressor cells (MDSC) found to rely upon IDO1 support are another key player in the establishment of an immunosuppressive tumor microenvironment. MDSC are an immature population of bone marrow-derived hematopoietic cells functionally defined by their ability to suppress T-cell activity [34]. In response to inflammatory signals, MDSCs migrate to the lymph node, spleen, and tumor tissue to create local immune suppression. Among the mechanisms utilized by MDSC to exert their T-cell suppressive effects [35–41], there is evidence that IDO1 activity is a critical factor. This connection was first revealed by genetic studies in IDO1−/− mouse models of de novo lung carcinoma and metastases [25]. IDO1−/− mice resisted the outgrowth of lung tumors, and MDSC obtained from tumor-bearing animals were impaired for suppression of CD8+ and CD4+ T cells. Moreover, IDO1 loss caused an attenuation of IL-6, a major driver of MDSC, and ectopic expression of IL-6 was sufficient to rescue impairment of the T-cell-suppressive activity of MDSC as well as the resistance to pulmonary metastasis in IDO1−/− mice [25]. Thus, IDO1 exerted regulatory control over MDSC suppressive function by its ability to influence the inflammatory milieu. Other studies show that IDO1 is needed for MDSC recruitment to tumors [42, 43]. In light of the pivotal role of IDO1 in supporting MDSC function, it is notable that no compelling evidence exists in mouse models that IDO1 is expressed directly in MDSC. In contrast, human studies have identified populations of IDO1-expressing MDSC and associated the IDO1 expression in those cells with immunosuppressive function [41, 44]. Overall, in addition to regulating MDSC function and recruitment, IDO1 may act through additional mechanisms to support MDSC activity.
17.4 IDO1 in Inflammatory Programming: Pathogenic Neovascularization and Metastasis
The critical importance of neoangiogenesis for supporting tumor outgrowth is well established [45]. Although angiogenesis is sometimes used to refer broadly to all blood vessel development, its specific meaning is the formation of new vessels from the pre-existing vascular network in contrast to vasculogenesis, which refers to vessel formation through recruitment of new cells such as bone marrow-derived endothelial precursor cells. While vasculogenesis has been predominantly associated with embryogenesis and angiogenesis with adult vessel formation, the picture is likely to be more complex, and the distinction between the two processes may not be absolute. A combination of vasculogenesis and angiogenesis has been implicated in the vascularization of organs of both mesodermal and endodermal origin such as the lung, heart, pancreas, and liver, while for organs of ectodermal origin, such as the brain, kidney, thymus, and limb bud, angiogenesis appears to be predominant [46]. Importantly, these observations suggest that the operative processes for forming new blood vessels may not be the same between different tissue environments, which may be a factor influencing the outgrowth of tumors and metastases at different sites in the body.
Neovascularization refers to the excessive and disorganized growth of blood vessels induced by ischemia in tissues such as the retina and lungs. Neovascularization is also a distinguishing characteristic of growing tumors. In experimental models of ischemia, immune cells have been reported to be important for pruning the excess vasculature and limiting neovascularization (Ishida, 2003) suggesting that immunity might play an important anti-neovascular role in tumors as well. In particular, the inflammatory cytokine IFNγ has been shown to trigger anti-neovascular activity that results in tumor cell killing. In a series of studies, IFNγ-mediated elimination of vessels was implicated as the primary mechanism for both CD4 and CD8 T-cell-dependent tumor rejection (Qin 2000, 2003). However, inflammation is a complex process that can also promote neovascularization. In particular, the inflammatory cytokine IL6 has been shown to be important for ischemia-induced neovascularization [47] and has been demonstrated to promote aberrant angiogenesis through a signaling process that does not require VEGF [48]. IL6 is also generally regarded as pro-tumorigenic as opposed to IFNγ, which is regarded as anti-tumorigenic. This raises the possibility that the cytokine balance in an inflammatory environment may influence tumor outgrowth by how it impacts neovascularization.
In this context, finding that loss of IDO1 resulted in diminished pulmonary vascularization [25] and suggested the hypothesis that the induction of IDO1 by IFNγ might be working in a negative feedback capacity to limit the anti-angiogenic impact of IFNγ and that this might be an important factor accounting for the ability of IDO1 to counteract immune-based restriction of tumor outgrowth. IDO1 loss was also associated with attenuated induction of the inflammatory cytokine IL6, and it was demonstrated in a pulmonary metastasis model that ectopic expression of IL6 could overcome the resistance to metastatic tumor outgrowth exhibited by IDO1−/− mice. These findings led to the hypothesis that IDO1 acts downstream of IFNγ and upstream of IL6 from the very onset of tumor initiation to shift the inflammatory environment toward angiogenesis and tumor promotion.
As predicted by this model, pulmonary metastases that developed in IDO1−/− mice exhibited significantly reduced neovascularization relative to their WT counterparts [49]. However, since overall metastatic tumor outgrowth in IDO1−/− mice was also significantly reduced, it was not clear if the reduction of blood vessel formation was a direct effect of IDO1 loss. To test the idea that IDO1 is important for supporting neovascularization outside other possible confounding effects within the tumor microenvironment, studies were conducted in a mouse OIR (oxygen-induced retinopathy) model, a well-established, reproducibly quantifiable surrogate system for studying neovascularization [50, 51]. As predicted, IDO1−/− mice exhibited a significant reduction in OIR-induced retinal neovascularization relative to their WT counterparts [49]. Loss of the related IDO2 isoform had no demonstrable effect on OIR-induced retinal neovascularization, indicating that the effect is specific to IDO1 [49]. No difference in the normal retinal vascularization that develops under normoxic conditions was observed between IDO1−/− and WT groups, and reduction of the avascular region [49], indicative of normal revascularization, was actually higher in the IDO1−/− animals indicative of an improvement in normal vascular regrowth occurring in mice lacking IDO1. The reduction in OIR-induced retinal neovascularization observed in mice lacking IDO1 genetically was recapitulated by siRNA-mediated knockdown of IDO1 expression in the retina [49], demonstrating that the effect of IDO1 loss on neovascularization could be elicited both locally and acutely. Likewise, pharmacologic inhibition of IDO1 with the clinical agent epacadostat reduced OIR-induced retinal neovascularization when delivered systemically to neonates [49]. In parallel studies, epacadostat administration in the pulmonary metastasis model resulted in rapid elimination of the existing neovasculature [49], validating the potential therapeutic relevance of these findings in the cancer setting.
Having established the importance of IDO1’s role in supporting neovascularization, studies were carried out to test the hypothesis that IDO1 produces this effect through its integration at the regulatory interface between the inflammatory cytokines IFNγ and IL6. Consistent with the hypothesis that IDO1 supports neovascularization primarily by counteracting the anti-angiogenic activity of IFNγ, the concurrent elimination of IFNγ in double knockout IFNG−/− IDO1−/− mice reverted the level of neovascularization in both the OIR and pulmonary metastasis models back to wild-type levels [49]. Conversely, IL6−/− mice, as predicted, exhibited a reduction in neovascularization in both the OIR and metastasis models similar to that observed in IDO1−/− mice [49]. The effect of IL6 loss on neovascularization was likewise reversed by the concomitant elimination of IFNγ in double knockout IFNG−/−IL6−/− mice [49], consistent with the hypothesis that the upstream potentiation of the pro-angiogenic activity of IL6 may be an important contributing factor in IDO1’s ability to support neovascularization. In all cases, neovascularization tracked closely with overall survival in the pulmonary metastasis model [49], indicating that the impact on tumor neovascularization may be a meaningful consequence of treatment IDO1 inhibitors that should be taken into consideration as part of the ongoing clinical development of these agents.
17.5 IDO2 in B-cell Inflamed States and Certain IDO1 Functions: Connections and Questions
Although relatively little studied as yet, IDO2 is a structural relative of IDO1 also implicated in modulating immunity through tryptophan catabolism, particularly autoimmunity [52]. The IDO2 gene is located immediately downstream of IDO1 in the mouse and human genomes, and structural studies suggest a more ancestral function for IDO2 [53]. Deletion of the IDO2 gene does not appreciably affect embryonic development, hematopoiesis, or immune character nor does it affect tryptophan or kynurenine levels in blood [54]. IDO2 enzyme activity clearly relies upon conditions that differ from IDO1, for example, in differing requirements for a physiological co-reductant system [55]. Indeed, earlier characterizations of the “weak” activity of IDO2 simply reflect nonoptimal biochemical conditions which when corrected confer demonstrable activity ([56]; L. Laury-Kleintop, J. DuHadaway and G.C. Prendergast, unpublished data). Thus, the lack of significant effects of IDO2 deletion on systemic blood levels in the mouse may reflect the far narrower normal range of IDO2 expression relative to IDO1 and TDO, which are relatively more broadly and strongly expressed.
Mouse genetic experiments establish a function for IDO2 in immunomodulation [52]. One notable feature of IDO2-deficient mice is a deficiency in their ability to support IDO1-induced T regulatory cells [54]. Parallel evidence of a similar tolerizing function for IDO2 in human dendritic cells has been reported [57]. IDO1-deficient mice have also been found to be mosaic deficient for IDO2 function, strengthening clues of IDO1-IDO2 interaction in immune control [54]. Figure 17.2 summarizes this feature of IDO2 and a model which captures its potential implications in cancer. Interestingly, in a model of autoimmune arthritis, indoximod (D-1MT) administration phenocopied the reduced disease severity associated with IDO2 deletion, and this therapeutic effect was abolished by IDO1 deletion [58], aligning with earlier evidence that indoximod can selectively disrupt IDO2 enzyme activity [59]. However, these connections may be contextual having yet to be extended in other systems (Fatokun, 2013 #4628 [60]), including humans where common genetic variations in IDO2 that reduce tryptophan catabolic activity may be relevant [59].
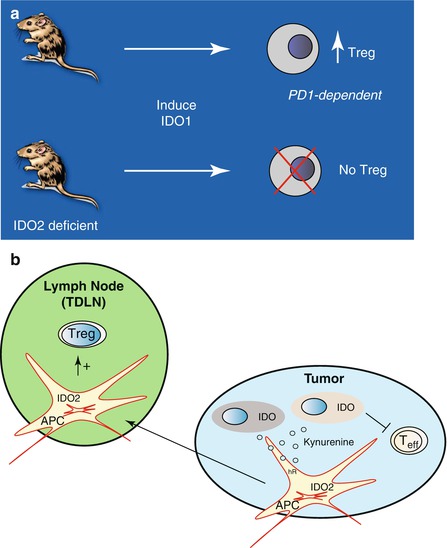
Fig. 17.2
IDO2 as a contributor to IDO1-mediated immune tolerance. (a) IDO2-deficient mice are defective in a PD1-dependent mechanism of IDO1-mediated Treg induction, in support of other evidence of IDO1-IDO2 genetic interaction in the mouse [54]. (b) Model. IDO2 expression activated by kynurenine/AhR signaling in APC acts to distally propagate tolerance signals produced locally by IDO1 in tumor and tumor-stromal cells (gray or pink cells in blue tumor). Local IDO1 expression blunts antigen-specific T effector cells mediated by kynurenine production and tryptophan deprivation [108]. IDO2 expression is upregulated in roving APC through the IDO1-mediated production of kynurenine, which acts through its receptor AhR to drive IDO2 transcription in APC [109]. IDO2 activity is licensed by IDO1 through transcriptional and posttranscriptional mechanisms [52]. APC are tolerized by kynurenine [110] and IDO2 is evoked as an effector in this model. IDO2 reinforces tolerance in APC by irreversible signals that differ from IDO1 signals which are reversible [59]. APC programmed by IDO2 rove to tumor-draining lymph nodes (green TDLN) or other metastatic sites where they reinforce IDO1-dependent Treg formation (IDO1 is also expressed in APC but not shown for clarity). This model is compatible with the latest model for IDO1 function in Treg formation [30], invoking IDO2 as a required intermediate function based on studies in IDO2-deficient mice [54]
Recent studies of the reduced susceptibility of IDO2−/− mice to autoimmune arthritis have revealed that IDO2 functions in B cells where it acts to support B-cell inflamed states [58, 61, 62]. These findings are interesting in light of evidence that certain cancers rely upon B-cell inflamed states for their development [63, 64]. While IDO2-deficient mice are unchanged with regard to their susceptibility to inflammatory skin carcinogenesis [54], they resist the development of K-Ras-induced pancreatic cancers (G.C. Prendergast and A.J. Muller, unpublished data). IDO2 enzymology differs from IDO1 in requiring different reductant systems, especially for the human enzymes, but recent elucidation of these differences confirms that IDO2 has demonstrable tryptophan catabolic activity [56]. While small-molecule inhibitors of mouse or human IDO2 have been reported [56, 65–68], they are not bioactive or for other reasons have not been studied in vivo as yet. Interestingly, a B-cell-penetrating bioactive antibody against IDO2 has been reported recently that phenocopies the anti-arthritic effects of IDO2-genetic deficiency in the mouse [62].
In normal tissues IDO2 expression is more narrow than IDO1 or TDO, being confined mainly to liver, kidney, brain, placenta, and antigen-presenting cells (APC) including B cells. Cancers do not tend to overexpress IDO2 although it has been reported in melanoma and gastric, brain, and pancreatic tumors, in the latter case rather widely [69]. IDO2 is regulated by the aryl hydrocarbon receptor (AhR) which binds kynurenine as an endogenous ligand produced by the more active IDO1 enzyme. Thus, given clues of IDO1-IDO2 interaction, it is conceivable that locoregional IDO1 activity may increase levels of IDO2 in roving antigen-presenting cells in the tumor microenvironment, perhaps contributing to a tolerized state that contributes to Treg formation in tumor-draining lymph nodes (TDLN). Figure 17.2 presents a model in which IDO2 functions on the Kyn effector pathway downstream of IDO1/TDO to positively modify decisions made in the TDLN to set tolerance to “altered-self” antigens, along the self-nonself continuum where immune challenges from autoimmunity and cancer arise [70].
17.6 Tryptophan Dioxygenase (TDO) in Inflammatory Programming: Immune Escape, Anoikis Resistance, and Metastasis
TDO expression in liver is responsible for homeostasis of tryptophan levels in the blood. Similar to IDO1, some tumors overexpress TDO as a means of immune escape [71–73]. Thus, there has been growing interest in small-molecule inhibitors of TDO as a parallel immunomodulatory strategy to attack tumors [74–79], the rationale for which has been reviewed in detail recently by pioneers in this area [72, 73]. The initial bioactive lead structure developed in the 1990s termed 68OC91 [74] has been used for mouse studies, but compounds optimized for potency and more favorable pharmacological profiles have been reported [75–78]. Deletion of the TDO-encoding gene TDO2 in the mouse causes higher concentrations of l-tryptophan to accumulate in blood, with some neurologic alterations perhaps attributable to a coordinate elevation in blood/brain levels of serotonin in these mice [80]. Interestingly, mice treated with the TDO2 inhibitor 680C91 will phenocopy TDO2−/− mice in showing an increased sensitivity to endotoxin-induced shock, implicating TDO in inflammatory programming [81]. However, despite this parallel with IDO1, as in the case with IDO2, there are differences in the inflammatory characteristics that appeared to be conferred by TDO, despite the common role of these enzymes in tryptophan catabolism [82]. While enzymological differences may help explain these different roles, it would also seem likely they reflect differences in locoregional control in the production of kynurenine and its metabolite or in the relative availability or efficiency of kynurenine effector mechanisms (AhR, kynurenine pathway catabolic enzymes, etc.). With regard to TDO, while there is evidence of its contribution to tumoral immune escape established preclinically with selective bioactive inhibitors [76, 83], neither a genetic proof in mice nor an understanding of the nature or extent of its expression in tumor cells or the tumor microenvironment has been established as yet. Moreover, TDO inhibitors pose different safety concerns from IDO1 inhibitors, including in the liver and central nervous system, as carefully discussed recently elsewhere [72]. That said, the rationale for developing TDO inhibitors as well as IDO-/TDO-combined inhibitors as next-generation modalities in the field continues to strengthen.
Recent emerging evidence suggests that TDO contributes to cancer-associated inflammatory programming like IDO1. Specifically, upregulation of TDO in cancer cells has been found to contribute to tumor cell survival and metastatic prowess beyond its role in immune escape [84]. Resistance to anoikis—a type of apoptosis triggered by cell adhesion deprival—is a key step in metastatic progression [28]. In a seminal study of aggressive “triple-negative” breast cancer (TNBC), D’Amato and colleagues showed how TDO upregulation in forced suspension culture was essential for anoikis resistance and metastatic capacity of TNBC cells [84]. Similar to its role in immune escape [83], kynurenine induction resulting from TDO upregulation was sufficient to activate the AhR signaling pathway, and pharmacological inhibition or genetic attenuation of TDO or AhR was each sufficient to restore anoikis sensitivity and reduce the invasive character of TNBC cells. Supporting these observations, tumor-bearing mice treated with the TDO inhibitor 680C91 exhibited reduced pulmonary metastasis. Lastly, elevated expression of TDO in clinical TNBC specimens was associated with increased disease grade, estrogen receptor-negative status, and shorter overall survival [84]. These findings extend the concept that TDO acts like IDO1 to drive a pathogenic inflammatory program(s) in cancer that extends beyond their contributions to enabling adaptive immune tolerance. Figure 17.3 summarizes ways in which IDO/TDO inhibitors may be used to leverage immune checkpoint therapy and chemotherapy through their effects on inflammatory programming and adaptive antitumor immune responses.
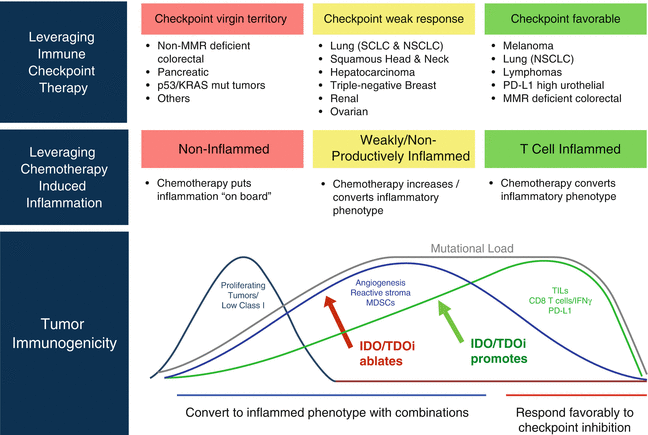
Fig. 17.3
IDO/TDO inhibitors to leverage immune checkpoint therapy and chemotherapy. IDO/TDO inhibitors are effective not only in combination therapeutic regimens, acting as immunomodulators to relieve immune escape and promote adaptive immune escape, but also to ablating or reprogramming inflammatory processes which can leverage the efficacy of chemotherapy as well as immune checkpoint therapy
17.7 Lead Clinical Agents: Indoximod, GDC-0919, and Epacadostat
17.7.1 Indoximod
We have published previously elsewhere a detailed discussion of the preclinical studies and rationale to embark upon clinical evaluation of this simple 1-methyl derivative of d-tryptophan [85, 86]. By far, the most commonly employed molecular probe to study IDO in the preclinical literature has been the d,l racemic mixture of 1-methyl-tryptophan (1MT). l-1MT is a weak substrate of IDO rather than a true inhibitor ( [52] #4610); D-1MT is neither a substrate nor an inhibitor of IDO, though in multiple model systems, it exhibits relatively greater antitumor properties associated with inhibition of IDO-mediated tryptophan catabolism in human dendritic cells [16]. Thus, neither are selective probes. As the first compound to enter phase I trials, indoximod was found to be well tolerated as a single agent or in combination with chemotherapy in studies which defined a dose of 1200 mg/day for ongoing evaluation in multiple phase II trials [87, 88]. Among this work, three notable trials focus on breast cancer patients in combination with Taxotere (chemotherapy combination), prostate cancer patients in combination with the dendritic cell vaccine sipuleucel-T (vaccine combination), and melanoma patients in combination with anti-PD1 (immune checkpoint combination) (http://clinicaltrials.gov/ct2/results?term=IDO&Search=Search; Vahanian et al., AACR 2017 late-breaking abstract). While the precise mechanism of action of indoximod has not yet been established definitively, striking cell-based experiments reveal that the mTORC1 pathway interprets indoximod at clinically relevant nanomolar concentrations as a mimetic of l-tryptophan [89]. Thus, indoximod may act in part by relieving the inhibitory effects of IDO-/TDO-mediated tryptophan deprivation on mTOR signals needed in T cells for antitumor activity. As further work reveals the precise mechanism of action, the low toxicity of indoximod as a simple d-tryptophan derivative remains an appealing feature of its clinical development, along with the opportunity it affords to leverage IDO1 and IDO/TDO enzymatic inhibitors.
17.7.2 GDC-0919
Prior to 2005, there was little motivation to develop inhibitors of IDO1, an unremarkable tryptophan-catabolizing enzyme. This situation changed with the first preclinical evidence of a role for IDO1 in cancer and of IDO1 inhibitor efficacy when combined with chemotherapy [8, 17–19]. In 2005 the only bioactive IDO inhibitor was 1-methyl-d,l-tryptophan (1-MT) with a reported K i of 34 μM [90, 91]. One of the few other reported IDO inhibitors at the time was 4-phenyl-imidazole (4-PI) identified in 1989 as a weak noncompetitive inhibitor of IDO1 by Sono and Cady [92]. Interestingly, although 4-PI showed noncompetitive inhibition kinetics through impressive spectroscopic studies, Sono and Cady showed that 4-PI was actually binding to the heme iron at the active site. Subsequently, the first crystal structure of IDO to be reported [93] confirmed this finding by showing 4-PI bound to the heme iron (Fig. 17.4a). This confirmation along with the rich crystal structure information facilitated the first structure-based drug-design activities of Malachowski and colleagues, seeding work in the phenylimidazole series from which the clinical lead GDC-0919 was later derived [21].
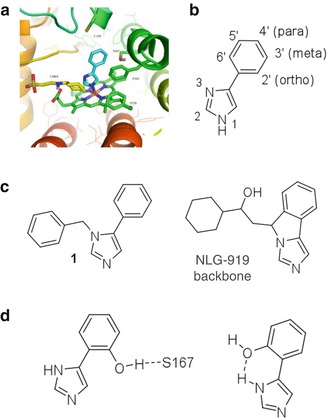
Fig. 17.4
Phenylimidazoles rooted in GDC-0919 development. (a) 4-PI bound to heme iron of IDO1. C129 is located above the 4-PI phenyl ring, while S167 resides in the back of the binding site. The buffer molecule CHES (yellow) is bound at the entrance of the active site of the IDO crystal structure. Graphics generated with PyMOL 1.0 [http://wwwpymol.org], an open-source molecular graphics system developed, supported, and maintained by DeLano Scientific LLC. http://www.delanoscientific.com. (b) Ring numbering of 4-phenylimidazole structures. (c) Structure of N-3 benzyl-substituted 4-PI and root structure of NLG-919. (d) Two possible benefits of 2′-OH substitution of 4-PI core
In early foundational work [21], Malachowski and colleagues explored 4-PI analogs to probe the active site of IDO1 with structural modifications that were focused on exploiting three binding interactions within the IDO active site: (1) the active site entrance region decorated with the heme 7-propionic acid, (2) the interior of the active site, in particular interactions with C129 and S167, and (3) the heme iron-binding group. The enhancement of IDO inhibition of 4-PI structures through interactions at the active site entrance focused on the N-1, C-2, and N-3 positions of the imidazole ring (Fig. 17.4b). All three positions were substituted with the goal of appending groups that would occupy the active site entrance. In the crystal structure of 4-PI with IDO, this region contains an N-cyclohexyl-2-aminoethanesulfonic acid (CHES) buffer molecule whose alkyl portion forms hydrophobic interactions with F163 and F226. In addition, the amino group of the CHES molecule forms an ion pair with the heme 7-propionic acid.
N-1 substituted 4-PI derivatives were completely devoid of inhibitory activity, which, not surprisingly, confirmed the binding of the N-1 nitrogen to the heme iron and, more importantly, demonstrated that the N-3 nitrogen of the imidazole cannot substitute to bind at the heme iron. However, N-3 benzyl-substituted derivatives (Fig. 17.4c1) were unexpectedly found to be roughly equipotent to 4-PI, thereby demonstrating that imidazole ring substitution was tolerable. The N-3 benzyl-substituted compound identified the correct imidazole ring location and spatial tolerance, likely occupying the active site entrance where the CHES buffer molecule sits in the IDO-4-PI crystal structure [93]. This discovery was consistent with the pharmacophore developed in studies of IDO1 inhibition by brassinin derivatives, i.e., a heme iron-binding group flanked by two large aromatic or hydrocarbon structures [94]. This proved prescient in light of subsequent development work by NewLink Genetics Inc. with regard to the backbone structure for the clinical candidate NLG-919 (Fig. 17.4c), renamed GDC-0919 after sublicense to Genentech/Roche. The NLG-919 backbone as shown extended from the same N-3 position to situate a similar hydrocarbon moiety in the active site entrance of IDO1.
Analysis of the crystal structure of 4-PI bound to IDO1 [93] indicated that S167 and C129 were in close proximity to the phenyl ring of 4-PI in the interior of the active site. Systematic evaluation of ortho, meta, and para substitutions of the phenyl ring with oxygen, sulfur, and fluorine was undertaken to ascertain if specific protein-ligand interactions could be exploited. The 2′-hydroxy (ortho substituted) modification afforded the most success generating a tenfold increase in potency relative to 4-PI (Fig. 17.4d). Two possibilities existed for this increased activity: intermolecular H-bonding with S167 or intramolecular H-bonding with N-3 to lock the phenyl and imidazole rings. The 2′,6′-dihydroxy-phenyl derivative, which presents a hydroxyl group to S167 or N-3 imidazole in either rotamer, was also synthesized, and it was roughly equipotent to the 2′-hydroxy derivative, thereby demonstrating that there was no additional benefit from both events. Subsequently, NewLink Genetics introduced a hydrocarbon bridge that replicates the H-bond to the N-3 imidazole, i.e., locking the conformation of the benzene and imidazole ring into one plane.
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


