Fig. 1.1
Clonal expansion of CML hematopoiesis. A reciprocal translocation between chromosomes 9 and 22 occurs at a hematopoietic stem cell (HSC) stage. This pre-leukemic stem cell expresses BCR-ABL at a low level and behaves like a normal HSC. To transform into an ultimate leukemia stem cell, the upregulation of BCR-ABL expression should be necessary. However, CML stem cells expressing high BCR-ABL transcripts are not necessarily addictive to the BCR-ABL kinase activity; thus, they are resistant to tyrosine kinase inhibitor (TKI) treatment. Instead, CML stem cells can survive by activating multiple BCR-ABL kinase-independent pathways. The massive proliferation of CML clone starts at a common myeloid progenitor (CMP) stage but not a stem cell stage. CMPs and their progeny are addictive to the BCR-ABL kinase activity for their survival and thus are sensitive to TKI treatment. It must be necessary to clarify the mechanisms of BCR-ABL upregulation in pre-leukemic stem cells to understand CML pathogenesis
1.5 CML Stem Cells Are Not Always Addicted to BCR-ABL Signaling
The constitutively active tyrosine kinase BCR-ABL is detectable in CML patients without exception. CML cells must be addicted to BCR-ABL signaling, because inhibition of BCR-ABL kinase activity by tyrosine kinase inhibitor (TKI) treatment dramatically reduced the leukemic burden of CML [31–33]. It is clear that proliferating CML progenitors are addicted to the BCR-ABL kinase activity. However, CML stem cells are not always addicted to BCR-ABL signaling because the majority of patients relapse after discontinuation of TKI treatment [34].
TKIs exert strong activity of kinase inhibition through binding to the kinase domain of BCR-ABL, and the vast majority of chronic-phase patients treated with TKIs achieve hematological and cytogenetic responses [31–33]. However, even in patients treated with TKIs for more than 5 years, minimal residual disease (MRD) is often detected by a highly sensitive PCR method despite the absence of TKI-resistant ABL mutations. Recently, several groups have shown that BCR-ABL-expressing cells persist in the CD34+CD38− human HSC fraction even after achievement of complete cytogenetic and molecular responses [35, 20, 27]. Of note, these cells retain a long-term repopulating capacity after xenotransplant into immunodeficient mice [20]. Based on these data, it is considered that TKIs are incapable of eradicating chronic-phase CML stem cells.
Recently, by using an inducible BCR-ABL transgenic mouse model, it has been demonstrated that CML stem cells can survive independent of BCR-ABL kinase activity [36]. In this model, p210 BCR-ABL expression is targeted to murine stem and progenitor cells via a tetracycline-off system. Upon tetracycline withdrawal, BCR-ABL expression is initiated at the HSC stage and a CML-like disease develops within a few weeks. Reintroduction of tetracycline completely blocked the BCR-ABL signaling and induced complete remission. However, CML-like disease was reconstituted from the remission marrow when tetracycline was stopped. This new mouse model formally proves that genetically induced CML stem cells can survive even if the expression of BCR-ABL oncogene was completely silenced for a certain period [36]. Thus, there might exist specific signaling pathways that support the survival of CML stem cells beyond the BCR-ABL kinase activity.
In clinics, in patients with complete molecular response (CMR) for more than 2 years, around 60 % of patients relapsed within 6 months after discontinuation of imatinib treatment [34]. The persistence of CML stem cells was observed even in patients treated with nilotinib, a second-generation strong TKI. At this moment, at most 50 % of chronic-phase CML patients achieve CMR and 40–50 % of these good responders successfully quit TKIs; thus, it is estimated that 75–80 % of chronic-phase CML patients need to continue TKI therapy throughout life. In order to “cure” CML, it is necessary to understand how CML stem cells survive under TKI treatment.
It is important to note that after the TKI discontinuation, a very low level of BCR-ABL transcripts remained detectable in a considerable fraction of patients who do not relapse. It is possible that these patients returned to the “pre-CML” phase in which CML stem cells are not addicted to BCR-ABL (Fig. 1.1). Another possibility is that some anti-leukemia immune responses inhibit CML stem cells to grow. Several groups have reported that treatment prior to TKI with interferon (IFN)-α is predictive of relapse-free survival upon the TKI discontinuation [37]. In this case, IFN-α is considered to target CML stem/progenitor cells as well as to facilitate an anti-leukemia immunity. These data suggest that combination of TKI and immune checkpoint therapies, such as anti-PD-1 or anti-PD-L1 antibody therapies, may be useful to eradicate CML stem cells.
1.6 Pathways That May Be Used for the Maintenance of CML Stem Cells
CML stem cells are likely to utilize key survival pathways that are inherent in normal HSCs. These pathways might also be good targets to eradicate CML stem cells. Previous studies have shown that Wnt/β-catenin [38] and Hedgehog [39] signaling pathways that are critical for normal HSC development and maintenance are also important for the maintenance of CML stem cells. Because inhibition of these pathways potentially influences the survival of normal HSCs, there is serious concern about whether the therapeutic window can be established appropriately in clinical trials. In addition, transcription factors such as Foxo family [40] and Hif1α [41] play critical roles in CML stem cell maintenance. BCL6 proto-oncogene was shown to be a key effector downstream of Foxo in self-renewal of CML stem cells [42]. Details are discussed in the following chapters.
It has been demonstrated that CSCs utilize the specific metabolic pathways. CML stem cells augment the expression of arachidonate 5-lipoxygenase (Alox5) which is responsible for producing leukotrienes, inflammatory substances [43]. The upregulation of Alox5 in CML stem cells occurs independent of BCR-ABL kinase activity. In the absence of Alox5, BCR-ABL transduction fails to induce a CML-like disease, and treatment with a 5-lipoxygenase inhibitor prolongs the survival of CML mice [43]. Importantly, normal HSCs are not affected by the inhibition of Alox5. Stearoyl-CoA desaturase 1 (Scd1), an endoplasmic reticulum enzyme that regulates fatty acid metabolism, is shown to be downregulated in CML stem cells [43]. Deletion of Scd1 gene accelerates the disease development in mouse CML model. Conversely, overexpression of Scd1 delays CML development, indicating that Scd1 might play a tumor-suppressive role [43]. Thus, the modulation of LSC-specific metabolism could also be useful to eradicate CML stem cells.
1.7 Conclusion
It has been considered that CML is a perfect model of oncogene addiction. Although proliferating CML progenitors are addictive to BCR-ABL kinase activity for their survival, most CML stem cells are resistant to TKI. To completely cure CML, it is necessary to fully understand the molecular events during development of CML stem cells from a single HSC that first acquires BCR-ABL fusion. In addition, elucidation of molecular events how CML stem cells survive during TKI therapy is critical. These studies are ongoing, and we are awaiting new drugs targeting such critical mechanisms.
References
1.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. doi:10.1038/35102167.CrossRefPubMed
2.
3.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. doi:10.1073/pnas.0530291100.PubMedCentralCrossRefPubMed
4.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–8.PubMed
5.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–7. doi:10.1158/0008-5472.can-06-2030.CrossRefPubMed
6.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–5. doi:10.1038/nature05384.CrossRefPubMed
7.
Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–35. doi:10.1016/j.cell.2005.03.032.CrossRefPubMed
8.
Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329(5991):568–71. doi:10.1126/science.1189992.PubMedCentralCrossRefPubMed
Related posts:
 Molecular Mechanisms of CML Stem Cell Maintenance
Recommendations for the Management of CML in the Era of Second-Generation TKIs
Molecular Mechanisms of CML Stem Cell Maintenance
Recommendations for the Management of CML in the Era of Second-Generation TKIs
 Goals of CML Treatment in the Tyrosine Kinase Inhibitor Era
Goals of CML Treatment in the Tyrosine Kinase Inhibitor Era
 Updated European LeukemiaNet Recommendations for the Management of CML
Updated European LeukemiaNet Recommendations for the Management of CML
 The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML
The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML
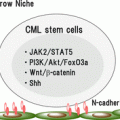 Roles for Signaling Molecules in the Growth and Survival of CML Cells
Roles for Signaling Molecules in the Growth and Survival of CML Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


