Hypothalamic—Pituitary Disease
Gerald Charnogursky
Nicholas Emanuele
Mary Ann Emanuele
Hong Lee
Shanika Samarasinghe
INTRODUCTION OF THE HYPOTHALAMIC—PITUITARY AXIS
The hypothalamic—pituitary unit is a complex neuroendocrine system that is responsible for secretion of anterior pituitary hormones, which are essential for reproduction, growth, metabolic homeostasis, responding to stress, and adapting to changes in the external environment. Each pituitary hormone is secreted in a distinctive pulsatile manner reflecting the interaction between hypothalamic neuroendocrine pathways, feedback effects from peripheral target gland hormones, and intrapituitary mechanisms.
Basal evaluation should consist of anterior pituitary and target organ hormone levels (Table 1.1). Further evaluation depends on the clinical question of hypofunctioning or hyperfunctioning of the suspected endocrine gland. Stimulatory tests provide insight into causes of hypofunction, whereas suppression tests are best if hyperfunction is suspected. Details of the specific evaluation methods of excess and deficiency of each hormone is described with each subsequent clinical entity.
PITUITARY ADENOMAS
Epidemiology and Pathogenesis
Pituitary adenomas can be classified based on size (<1 cm are microadenomas and ≥1 cm are macroadenomas) or functionality (nonsecretory or secretory). Most pituitary tumors are microadenomas and many are nonfunctioning. The prevalence of pituitary adenomas at autopsy is as high as 20% to 25% [1]. The majority of pituitary adenomas occur sporadically. While the most common cause of a pituitary mass is an adenoma, the differential diagnosis includes infiltrative processes, infections, lymphocytic hypophysitis,
craniopharyngioma, cysts, empty sella, benign and malignant tumors, and metastatic carcinoma [2].
craniopharyngioma, cysts, empty sella, benign and malignant tumors, and metastatic carcinoma [2].
Table 1.1. Pituitary and Target Hormones | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Patients with pituitary adenomas can be asymptomatic or present with symptoms due to mass effect, pituitary hormone dysfunction, or both [2]. Nonfunctioning intrasellar adenomas are often asymptomatic and usually diagnosed incidentally on imaging. The magnitude and severity of symptoms depend on several factors including size, location, and rate of enlargement. In hormone-secreting tumors with associated clinical syndromes, an appropriate hormone assessment will determine the best initial treatment and the need for further therapy. Surgical, pharmacologic, and radiotherapeutic treatments are available for hormone-secreting pituitary tumors.
Patients with large or rapidly enlarging tumors or those with rapid expansion of the contents of the sella have acute onset of signs and symptoms associated with neurologic and/or hormonal dysfunction. This is classically seen in pituitary apoplexy when there is acute hemorrhage or infarction of a preexisting pituitary adenoma (usually a macroadenoma). Patients commonly present with sudden onset of a severe headache, which is usually located in the retro-orbital region. Other symptoms may include nausea, lethargy, altered consciousness, cranial nerve palsies, and visual field defects. Compression of the pituitary gland and its vascular supply can lead to partial or complete hypopituitarism.
Diagnosis
Subsequent sections of this chapter discuss the diagnosis of hyperprolactinemia, acromegaly, Cushing disease (CD), and thyroid-stimulating hormone (TSH)-producing and gonadotropin-producing pituitary tumors. A sellar mass without excess hormone production indicates a nonfunctional pituitary adenoma (NFPA). The α-subunit, common to follicle-stimulating hormone (FSH), luteinizing hormone (LH), and TSH and lacking metabolic activity, can be elevated in nonfunctioning adenomas. If initially high, this level can be followed as a marker of tumor recurrence. The β-subunit of these glycoproteins is unique and confers specificity to the hormone.
Treatment
Overview
Tumor type, function, and size determine therapy for pituitary tumors. Initial evaluation should assess the presence and type of hormone hypersecretion, any
hormonal deficiencies and the need for replacement therapy, any visual abnormality, and extrasellar extension. Therapeutic interventions include medical therapy, transsphenoidal or frontal surgery, and radiotherapy. Details of pharmacologic treatment for specific lesions are discussed later in this chapter. Transsphenoidal surgery is indicated in most patients with tumors that secrete growth hormone (GH), adrenocorticotropic hormone (ACTH), and TSH and with large nonfunctional tumors. Radiotherapy, conventional external beam or stereotactic, can benefit patients with significant residual tumor in whom medical and surgical therapy have been unsuccessful. Ideally, excess levels of pituitary hormones normalize, and the hypothalamic—pituitary-target-organ responses, both stimulatory and inhibitory, are restored. Other goals of therapy are relief of headaches and reversal of visual loss.
hormonal deficiencies and the need for replacement therapy, any visual abnormality, and extrasellar extension. Therapeutic interventions include medical therapy, transsphenoidal or frontal surgery, and radiotherapy. Details of pharmacologic treatment for specific lesions are discussed later in this chapter. Transsphenoidal surgery is indicated in most patients with tumors that secrete growth hormone (GH), adrenocorticotropic hormone (ACTH), and TSH and with large nonfunctional tumors. Radiotherapy, conventional external beam or stereotactic, can benefit patients with significant residual tumor in whom medical and surgical therapy have been unsuccessful. Ideally, excess levels of pituitary hormones normalize, and the hypothalamic—pituitary-target-organ responses, both stimulatory and inhibitory, are restored. Other goals of therapy are relief of headaches and reversal of visual loss.
Surgery
Surgery remains the first-line treatment for symptomatic pituitary tumors with the exception of prolactinomas. Indications include mass effect with visual compromise, primary correction of hormonal hypersecretion, resistance to medical therapy, pituitary hemorrhage/apoplexy, and need for tissue for diagnosis. The most rapid and reliable relief from optic nerve and chiasmal compression is surgery; visual deficits improve in over 85% of patients [3]. Minimally invasive transsphenoidal microsurgery is used in the vast majority of pituitary tumors. This approach precludes invasion of the cranial cavity and manipulation of the brain [4]. Normal pituitary tissue can be clearly distinguished from tumor tissue, facilitating dissection. The mortality in experienced hands is low, between 0.2% and 1% [4]. A transcranial approach is undertaken in tumors with significant temporal or suprasellar extension, especially those contained by a small diaphragmatic aperture (dumbbell configuration), invading into the posterior clivus, or coexisting with intrasellar vascular lesions. An endoscopic approach is increasingly being utilized. Use of an endoscope allows for a superior view of the target area, an enlarged working angle with panoramic views of bony landmarks, and access to tumor extension into the cavernous sinus. Disadvantages include management of perioperative intrasellar bleeding and the requirement for a preoperative computed tomography (CT) scan [3]. Additional advances in the operating room include intraoperative magnetic resonance imaging (MRI) which allows for real-time assessment of the dimensions and extent of the pituitary mass concealed from the operating microscope. Neuronavigation employs a 3D data set provided by preoperative imaging to gain additional anatomical orientation, identifying tumor shape and major arteries superimposed onto the surgical field, and improves the rate of completely excised tumors [4].
In terms of surgical outcomes, the tumor size, magnitude of hormonal hypersecretion, local invasion, previous therapy, and experience of the neurosurgeon are critical factors in determining the likelihood of success. In hormonally active pituitary microadenomas, expert surgeons report remissions in up to 90% of patients [3]. However, with macroadenomas, remission rates vary widely depending on the series and definition of cure.
Because most pituitary tumors are benign, the results of surgery are usually gratifying, particularly in patients with suprasellar extension and visual abnormalities. Improvement in visual field abnormalities occurs in 80% of such patients, progression of visual disturbance is arrested in 16%, whereas visual deterioration occurs in 4% [1]. Prevention of recurrence and avoidance of diabetes insipidus (DI) are perhaps the most challenging goals of pituitary tumor surgery. Surgery provides a complete histopathologic characterization of the lesion. A tissue diagnosis is desirable, since the differential diagnosis of sellar masses
is wide and some lesions can present as pseudoprolactinomas (hyperprolactinemia secondary to pituitary stalk compression or hypothalamic damage, which leads to interference with dopaminergic inhibition of prolactin [PRL] secretion). Craniopharyngiomas demonstrate variable tumor extension; thus, total resection is more difficult to achieve and associated with higher postoperative morbidity.
is wide and some lesions can present as pseudoprolactinomas (hyperprolactinemia secondary to pituitary stalk compression or hypothalamic damage, which leads to interference with dopaminergic inhibition of prolactin [PRL] secretion). Craniopharyngiomas demonstrate variable tumor extension; thus, total resection is more difficult to achieve and associated with higher postoperative morbidity.
Postoperative Complications
The low morbidity and mortality associated with transsphenoidal surgery is a major advantage of this approach. Anterior and posterior pituitary dysfunction are assessed after transsphenoidal surgery. Disruption of the hypothalamic— pituitary—adrenal axis (HPA) is covered with perioperative glucocorticoids. Anterior pituitary insufficiency and DI can occur in up to 20% of cases [5]. Monitoring for water imbalances due to deficiency or excess of arginine vasopressin (AVP) requires strict assessment of fluid intake, urine output, and urine osmolality in conjunction with frequent electrolyte measurements. DI is often transient in the postoperative period. Most patients maintain adequate fluid intake and normal volume status. Desmopressin (DDAVP) therapy should be given with caution and is usually only needed briefly. If the patient is alert with intact thirst mechanisms, thirst can be used to guide water replacement. An analysis of 1,571 patients undergoing transsphenoidal surgery demonstrated various patterns of postoperative polyuria and hyponatremia; only 0.9% of patients at 3 months and only 0.25% of patients at 1 year still had polyuria or needed DDAVP [5].
Major neurosurgical complications, including cerebrospinal fluid leak, meningitis, stroke, intracranial hemorrhage, and visual loss are relatively rare (1.5%—4%). Minor complications including sinus disease, nasal septal perforation, epistaxis, and wound issues occur in approximately 6.5% [8]. As transcranial surgery necessitates direct dissection of the brain, vascular structures, and visual pathways, the complication rate is considerably higher.
Mortality rates of 0.86%, 0.27%, and 2.5% have been reported in patients with macroadenomas, microadenomas, and macroadenomas previously treated by other modalities, respectively [4, 5, 6, 7]. For patients with previous treatment or macroadenomas, visual loss occurred in 2.5% and 0.1%, leakage of cerebrospinal fluid in 5.7% and 1.3%, stroke or vascular injury in 1.3% and 0.2%, meningitis or abscess in 1.3% and 0.1%, and oculomotor palsy in 0.6% and 0.1%, respectively. The incidence of postoperative hypopituitarism is about 3% in patients with microadenomas, and this increases slightly with size and invasiveness of the tumor. The background rate of pituitary tumor recurrence is about 1% to 2% per year in patients treated by surgery alone; postoperative external pituitary irradiation reduces this level. More recent studies have emphasized the effectiveness of administering radiotherapy after initial surgery to reduce the risk of tumor regrowth in patients with residual mass or hormone hypersecretion [8].
Radiation
Radiotherapy is reserved for patients who have failed surgical and/or medical therapies or who are unable to tolerate surgery due to medical comorbidities. This includes those with persistent or recurrent hormonal hypersecretion states. It is the rational step after failed transsphenoidal surgery in CD as medical therapy is poor. Pituitary irradiation has also decreased the incidence of Nelson syndrome, locally aggressive ACTH-producing tumors due to loss of feedback inhibition at the anterior pituitary ACTH secreting corticotrope in patients who have undergone bilateral adrenalectomy. Postoperative radiotherapy is favored for craniopharyngioma [9].
Radiotherapy has a high therapeutic index, delivering high-energy ionizing radiation to arrest growth of tumor while minimizing exposure to normal
surrounding tissue [10]. The aim is to prevent tumor progression and normalize elevated hormone levels. It is successful in achieving tumor control in up to 95% to 97% of patients at 10 years and 88% to 92% of patients at 20 years. Hormonal control is less successful with 60% to 80% of patients in remission, depending on tumor type after radiotherapy. However, prompt reduction in either tumor size or hormone hypersecretion is rare. Reduction in hormone hypersecretion may occur within 3 to 6 months of therapy, but attainment of normal values usually requires at least 5 and often 10 years [11]. Hypopituitarism is the most common complication, with up to 80% of patients demonstrating gonadotroph, somatotroph, thyrotroph, or corticotroph deficits within 10 years after radiation [12]. In one study, half the patients treated with conventional supervoltage radiation developed hypopituitarism within 26 months of therapy [13]. In another series, at least one-third of patients had pituitary deficiencies within 2 to 3 years [14]. The incidence of hypopituitarism increases with length of follow-up, necessitating lifelong monitoring of patients with appropriate hormone measurements and dynamic studies. Other complications of radiotherapy include optic neuropathy (1.5% risk), brain necrosis (0.2%), secondary malignancies such as gliomas and astrocytomas (2%), cerebrovascular disease, and possible cognitive impairment [12]. The development of modern stereotactic technologies that can deliver high-dose radiation more precisely to the tumor can minimize complications and allow for more rapid reduction in hormonal hypersecretion. Stereotactic irradiation can be given as a single-fraction radio surgery using either a cobalt unit (gamma knife) or linear accelerator (LINAC) or as lower fractionated doses over several treatments using a LINAC or proton source [8]. The precision achieved with stereotactic techniques allows for sparing of the normal brain from high radiation doses. The type of radiation treatment administered must be individualized according to the tumor location (proximity to the optic chiasm and cavernous sinus) and the availability of the radiation source. Despite theoretical advantages, long-term outcomes and efficacy have yet to be defined for stereotactic radiation.
surrounding tissue [10]. The aim is to prevent tumor progression and normalize elevated hormone levels. It is successful in achieving tumor control in up to 95% to 97% of patients at 10 years and 88% to 92% of patients at 20 years. Hormonal control is less successful with 60% to 80% of patients in remission, depending on tumor type after radiotherapy. However, prompt reduction in either tumor size or hormone hypersecretion is rare. Reduction in hormone hypersecretion may occur within 3 to 6 months of therapy, but attainment of normal values usually requires at least 5 and often 10 years [11]. Hypopituitarism is the most common complication, with up to 80% of patients demonstrating gonadotroph, somatotroph, thyrotroph, or corticotroph deficits within 10 years after radiation [12]. In one study, half the patients treated with conventional supervoltage radiation developed hypopituitarism within 26 months of therapy [13]. In another series, at least one-third of patients had pituitary deficiencies within 2 to 3 years [14]. The incidence of hypopituitarism increases with length of follow-up, necessitating lifelong monitoring of patients with appropriate hormone measurements and dynamic studies. Other complications of radiotherapy include optic neuropathy (1.5% risk), brain necrosis (0.2%), secondary malignancies such as gliomas and astrocytomas (2%), cerebrovascular disease, and possible cognitive impairment [12]. The development of modern stereotactic technologies that can deliver high-dose radiation more precisely to the tumor can minimize complications and allow for more rapid reduction in hormonal hypersecretion. Stereotactic irradiation can be given as a single-fraction radio surgery using either a cobalt unit (gamma knife) or linear accelerator (LINAC) or as lower fractionated doses over several treatments using a LINAC or proton source [8]. The precision achieved with stereotactic techniques allows for sparing of the normal brain from high radiation doses. The type of radiation treatment administered must be individualized according to the tumor location (proximity to the optic chiasm and cavernous sinus) and the availability of the radiation source. Despite theoretical advantages, long-term outcomes and efficacy have yet to be defined for stereotactic radiation.
HYPOPITUITARISM
Mary Ann Emanuele
Nicholas Emanuele
Pathophysiology
Total or partial hypopituitarism may occur in patients with pituitary adenomas, parasellar invasion, or hypophysitis; after pituitary surgery or radiation (including head and neck radiation for malignancy); or after head injury [15, 16]. Pituitary apoplexy resulting from bleeding into an existing adenoma is also commonly associated with hypopituitarism [17]. This can occur postpartum when the pituitary is markedly enlarged, after a complicated delivery with excessive bleeding and hypotension (Sheehan syndrome). In a retrospective 20-year chart review of patients with classical pituitary apoplexy, approximately 90% had permanent hypopituitarism, independent of surgical decompression or not [18]. Other causes of hypopituitarism include compression of normal pituitary tissue (empty sella, suprasellar masses, metastases, and internal carotid artery aneurysm) and infiltrative processes like leukemia, granulomatous disease, or histiocytosis X. Congenital deficiencies of pituitary hormones due to genetic defects can lead to isolated or combined hormone deficiencies and at times are associated with specific syndromes [18].
Deficiency of any or all of the six major hormones (LH, FSH, GH, thyrotropin, corticotropin, and PRL) can occur. The clinical picture will depend on the type, degree, and rapidity of onset of pituitary hormone deficiency (PHD). Most of the
signs and symptoms of PHDs are similar to the hormone deficiencies associated with the target gland failure. In patients with pituitary macroadenomas, 30% or more can have PHDs, with the most common being growth hormone deficiency (GHD). In patients with microadenomas, gonadotropin deficiency is the most common. PRL deficiency is uncommon except with pituitary infarction.
signs and symptoms of PHDs are similar to the hormone deficiencies associated with the target gland failure. In patients with pituitary macroadenomas, 30% or more can have PHDs, with the most common being growth hormone deficiency (GHD). In patients with microadenomas, gonadotropin deficiency is the most common. PRL deficiency is uncommon except with pituitary infarction.
Clinical Findings
Secondary Adrenal Insufficiency
Clinical Presentation
Patients with secondary adrenal insufficiency can present with a wide range of symptoms depending on the degree and duration of ACTH and consequent glucocorticoid deficiency. The symptoms can range from subtle nonspecific symptoms to hypotension and adrenal crisis. Excluding adrenal crisis, patients with secondary adrenal insufficiency commonly report fatigue, weakness, mental dysfunction, dizziness (orthostatic hypotension may be present), anorexia, and weight loss. Women may have decreased libido and lose pubic and axillary hair since the adrenals are the major source of androgen in females. In males, beard and body hair are preserved unless gonadotropin deficiency coexists. Patients with secondary adrenal insufficiency who are not on glucocorticoid therapy are at high risk for adrenal crisis when under increased stress (major medical illness, surgery, etc.) since the pituitary—adrenal axis may not be capable of responding appropriately due to diminished ACTH reserve. Patients with adrenal crisis can present with nausea, vomiting, abdominal pain, severe hypotension, or hypovolemic shock. These patients do not have hyperpigmentation, in contrast to patients with primary adrenal insufficiency (Addison disease) who have hyperpigmentation due to elevated proopiomelanocortin (POMC) and ACTH levels.
Biochemical Diagnosis
Patients have a low cortisol level with low or inappropriately normal ACTH level. Patients usually have normal sodium levels but may have hyponatremia. Cortisol suppresses AVP. With cortisol deficiency, lack of negative feedback on vasopressin secretion allows for increased AVP levels, leading to retention of free water and hyponatremia. Long-term ACTH deficiency leads to atrophy of all zones of the adrenal cortex including the zona glomerulosa, which makes aldosterone. This lack of aldosterone further intensifies hyponatremia. Serum potassium concentration is usually normal in secondary adrenal insufficiency as the renin—angiotensin—aldosterone system is intact (in contrast to primary adrenal insufficiency where there is mineralocorticoid deficiency). However, with long-term ACTH deficiency, atrophy of the entire adrenal cortex can occur with subsequent hyperkalemia. Metabolic acidosis, hypercalcemia, hypoglycemia, normocytic anemia, and eosinophilia may also be present [19].
Measurement of ACTH is required. ACTH is secreted in a pulsatile fashion with a circadian rhythm and increased during stress. ACTH results must be interpreted with knowledge of collection time, clinical stress level, and whether exogenous synthetic glucocorticoids were previously administered. A simultaneous plasma cortisol sample is necessary to interpret the appropriateness of the plasma corticotropin level. An 8 a.m. cortisol between 10 and 20 mg/dl effectively excludes adrenal insufficiency, although patients with secondary (pituitary dysfunction) or tertiary (hypothalamic dysfunction) cortisol deficiency may have low-normal plasma cortisol levels (<3 ng/ml). If clinical suspicion is high, further evaluation with provocative testing should be pursued.
The gold standard for diagnosing secondary adrenal insufficiency is the insulin hypoglycemia test. However, in the acute setting, treatment of patients with
high likelihood of secondary adrenal insufficiency based on history and clinical assessment should not be delayed. Patients can be treated with glucocorticoids, and further testing can be done at a later time [19]. The insulin-induced hypoglycemia test is performed in the morning after an overnight fast. After baseline glucose, ACTH, and cortisol levels are drawn, intravenous regular insulin is given IV push (0.1-0.15 U/kg), and serum (or blood) glucose, ACTH, and cortisol levels are measured at 15, 30, 60, 90, and 120 minutes after the initial bolus. If there is no laboratory or clinical evidence of hypoglycemia (e.g., tachycardia and sweating) after 45 minutes, the insulin should be rebolused and sampling repeated. Adequate hypoglycemia occurs when the blood glucose drops below 40 mg/dl. Clinical manifestations of hypoglycemia and a plasma glucose level below 40 mg/dl are required for the interpretation of ACTH and cortisol levels. If these two criteria are fulfilled, the plasma cortisol level should exceed 20 mg/dl. If this level of cortisol is achieved, the HPA is intact and the clinical syndrome being evaluated is not due to HPA disease. If not achieved, and the basal ACTH level was not elevated, then the syndrome is due to HPA dysfunction. Ischemic heart disease, cardiac conduction disorders, and epilepsy are contraindications to the test.
high likelihood of secondary adrenal insufficiency based on history and clinical assessment should not be delayed. Patients can be treated with glucocorticoids, and further testing can be done at a later time [19]. The insulin-induced hypoglycemia test is performed in the morning after an overnight fast. After baseline glucose, ACTH, and cortisol levels are drawn, intravenous regular insulin is given IV push (0.1-0.15 U/kg), and serum (or blood) glucose, ACTH, and cortisol levels are measured at 15, 30, 60, 90, and 120 minutes after the initial bolus. If there is no laboratory or clinical evidence of hypoglycemia (e.g., tachycardia and sweating) after 45 minutes, the insulin should be rebolused and sampling repeated. Adequate hypoglycemia occurs when the blood glucose drops below 40 mg/dl. Clinical manifestations of hypoglycemia and a plasma glucose level below 40 mg/dl are required for the interpretation of ACTH and cortisol levels. If these two criteria are fulfilled, the plasma cortisol level should exceed 20 mg/dl. If this level of cortisol is achieved, the HPA is intact and the clinical syndrome being evaluated is not due to HPA disease. If not achieved, and the basal ACTH level was not elevated, then the syndrome is due to HPA dysfunction. Ischemic heart disease, cardiac conduction disorders, and epilepsy are contraindications to the test.
The corticotropin-releasing hormone (CRH) stimulation test can be used to distinguish between hypothalamic and pituitary causes of secondary hypoad-renalism. An intravenous bolus of synthetic ovine CRH (1 mg/kg body weight or 100 μg total dose) is injected after the patient fasts for a minimum of 4 hours. Blood samples are collected 15 and 0 minutes before and 5, 10, 15, 30, 45, 60, 90, and 120 minutes after CRH injection and analyzed for ACTH and cortisol. A normal response is documented when plasma ACTH increases by at least 35% and/or the serum cortisol increases by at least 20%. Attenuated plasma ACTH and serum cortisol responses to CRH are seen in patients with pituitary ACTH deficiency. Those with hypothalamic disease have exaggerated and prolonged plasma ACTH response and a subnormal cortisol response.
The cosyntropin (synthetic ACTH 1-24) stimulation test bypasses the hypothalamus and pituitary and directly stimulates the adrenals. Therefore, in the strictest sense, this is an adrenal stimulation test. This test and its interpretation are described under the adrenal gland chapter.
An alternative method for testing the HPA axis is the metyrapone test. Metyrapone blocks synthesis of cortisol by blocking 11β-hydroxylase. The induced cortisol deficiency stimulates ACTH production in normal individuals. This ACTH increase leads to an increase in the precursor to cortisol, 11-deoxy-cortisol (compound S), thus testing the ACTH reserve. Metyrapone 750 mg every 4 hours is given orally for 24 hours. In unaffected patients, it suppresses the 8 a.m. cortisol level to less than 7 mg/dl and increases the serum 11-deoxycortisol greater than or equal to 10 mg/dl. In patients with hypothalamic or pituitary disease, the serum 11-deoxycortisol level is below 10 mg/dl. Hydrocortisone 100 mg should be given intravenously to reverse the cortisol deficiency after the 8 a.m. samples are taken. A shorter version of this test involves the administration of a single metyrapone 750-mg dose at midnight and measurements of serum 11-deoxycortisol and cortisol at 8 a.m. However, this shorter test may not separate normal from abnormal as well as the longer test.
Treatment
Treatment of cortisol deficiency is described in the chapter on the adrenal gland. Every patient receiving adrenal replacement therapy should wear an identifying necklace or bracelet to alert caregivers in the event of medical emergency.
Central or Secondary Hypothyroidism
Clinical Presentation
Secondary or central hypothyroidism (CH) is a rare cause of hypothyroidism characterized by deficiency of TSH. Both primary and secondary hypothyroidism can manifest with the same clinical picture. In CH, symptoms of hypothyroidism tend to be milder than in primary hypothyroidism since there is often some residual TSH secretion [20]. Patients can present with fatigue, cold intolerance, constipation, weight gain or inability to lose weight, dry skin, hair loss, puffiness, nonpitting edema (myxedema), diffuse myalgias exacerbated by exercise, carpal tunnel syndrome, peripheral neuropathy, cognitive impairment, and rarely myxedema coma. On examination, bradycardia and diastolic hypertension (because of loss of the vasodilatory effects of thyroid hormone) can be present, as well as periorbital edema, proximal muscle weakness, and a slow relaxation phase of reflexes. Patients with secondary hypothyroidism do not present with goiter in contrast to those with primary hyperthyroidism [20].
Biochemical Diagnosis
Patients have a low free and total triiodothyronine and thyroxine levels with inappropriately normal or low TSH. If the thyrotropin concentration is normal in association with normal serum thyroid hormone levels, the patient is euthyroid and does not require further testing. If the serum thyroid hormone levels are low and the thyrotropin level is normal (but inappropriately low for the prevailing thyroid hormone levels) or is low, the patient has secondary (from pituitary disease) or tertiary (from hypothalamic dysfunction) hypothyroidism. In the past, pituitary versus hypothalamic failure could be distinguished by administering thyrotropin-releasing hormone (TRH), but TRH is no longer available. Most patients also suffer from other PHDs [26]. CH may be masked by GHD. In one study, 36% of patients with GHD subsequently needed thyroid hormone replacement after GH replacement; the thyroid deficiency was not recognized until after the GH replacement [21].
Treatment
This deficiency is treated with oral levothyroxine 0.075 to 0.15 mg once daily or 1.67 μg/kg body weight daily. The dose is adjusted according to the clinical response, and the serum free thyroxine (free T4) values should be in the middle to upper part of the normal range. Measurement of serum TSH is of no value in assessing a response to levothyroxine in patients with hypothalamic—pituitary disease.
Secondary Hypogonadism
Clinical Presentation
Gonadotropin deficiency, also termed hypogonadotropic hypogonadism, results from either a pituitary defect or a deficiency of hypothalamic gonadotropin-releasing-hormone (GnRH). Acquired causes include acute or chronic illness. The illness may be medical or psychiatric like anorexia nervosa. Acute illness may lead to transient secondary hypogonadism that may reverse weeks to months after the illness resolves. Other causes include use of long-acting GnRH analogs or opiates, which can suppress gonadotropins and long-term use of glucocorticoids [22]. Hyperprolactinemia can affect the GnRH pulse generator and therefore lower LH/FSH production causing hypogonadism. In women, Sheehan syndrome— pituitary infarct due to peri/postpartum hemorrhage—can lead to hypogonadotropic hypogonadism [23].
Secondary hypogonadism can also be congenital. An example of congenital hypogonadotropic hypogonadism is Kallmann syndrome wherein GnRH neurons fail to migrate from the olfactory placode into the hypothalamus during fetal life. Therefore, GnRH is not produced by the hypothalamus, and the pituitary gland is not stimulated to make LH/FSH. Kallmann syndrome can be sporadic or familial (X-linked, autosomal dominant or recessive). Anosmia, midline facial abnormalities, urogenital abnormalities, and synkinesis also can be seen in this disorder [23].
Gonadotropin deficiency often occurs early in the course of hypopituitarism. Hypogonadism is often diagnosed retrospectively after the patient exhibits the symptoms of a mass lesion.
Clinical manifestations of secondary hypogonadism vary, depending on the age of onset. If hypogonadism is present before puberty in men, it can lead to delayed puberty, presence of small testes, small phallus, scant pubic and axillary hair, as well as eunuchoid proportions (disproportionately long arms and legs secondary to delayed epiphyseal closure). There is reduced male musculature, gynecomastia, and persistent high-pitched voice. Men who develop hypogonadism after puberty can present with decreased libido, erectile dysfunction, hot flashes (less common), diminished facial and body hair, fine facial wrinkles, gynecomastia, soft and small testes, progressive decrease in muscle mass, increase in visceral fat, oligo/azoospermia and decreased bone mass [23]. In women, if hypogonadism presents prior to puberty, there can be primary amenorrhea and absent breast development. After puberty, premenopausal women can present with menstrual irregularities, hot flashes, vaginal dryness, dyspareunia, breast atrophy, decreased libido, and low bone density [22].
Biochemical Diagnosis
Gonadotropin deficiency is diagnosed by measuring basal serum LH, FSH, and concomitant gonadal steroids (free testosterone in men and estradiol in women). A low free testosterone or estradiol with low or inappropriately normal levels of gonadotropins is suggestive of hypothalamic—pituitary dysfunction. Usually, dynamic tests are not needed for diagnosis. In males, serum testosterone has a diurnal variation and is higher in the early morning; therefore, blood must be drawn at about 8 a.m. Even though LH and FSH are secreted in a pulsatile fashion in men, levels of these hormones fall within a fairly narrow range. The LH and FSH levels should be interpreted with the clinical findings, simultaneous testosterone level, and possibly semen analysis. In women, marked changes in gonadotropin secretion occur during different phases of the menstrual cycle. Measurement of LH and FSH in a woman who is not taking oral contraceptives and who has regular menstrual cycles is usually not indicated since gonadotropin deficiency is not likely in this clinical setting. Documentation of a normal menstrual cycle and normal luteal-phase serum progesterone level excludes significant gonadotropin dysfunction. In amenorrheic or oligoamenorrhic nonpregnant women, measurement of serum LH, FSH, estradiol, and PRL levels can provide insight into the cause of hypogonadism. Primary ovarian failure presents with increased LH and FSH, whereas hypogonadism due to pituitary or hypothalamic causes have low estradiol and low or inappropriately normal LH and FSH levels.
Treatment
Treatment of hypogonadism is described in the chapter on reproduction.
Growth Hormone Deficiency
Clinical Presentation
GHD in children results in growth failure. However, symptoms of GHD in adults are nonspecific, and evaluation for this disorder is usually performed in adults
with a history of GHD in childhood or known pituitary disease. GHD is likely if the patient has documented deficiency of other pituitary hormones including TSH, ACTH, and gonadotropins [1]. In adulthood, GHD can present with reduced energy and depressed mood. GH-deficient subjects have a diminished sense of physical and psychological well-being (e.g., less energy, emotional lability, sense of social isolation, and diminished libido). GH lack contributes to decreased lean body mass and increased abdominal adiposity. Exercise capacity is impaired and there is decreased muscle mass and muscle strength. The contractile properties of muscles are affected in patients with GHD [24]. Fracture risk is increased since bone density, particularly in the lumbar spine, is reduced in some patients with adult-onset GHD. Levels of serum low-density-lipoprotein (LDL) cholesterol may be increased, whereas high-density-lipoprotein (HDL) cholesterol remains normal. Deficiency of GH may contribute to the increased cardiovascular mortality found in patients with hypopituitarism who receive replacement hormones other than GH.
with a history of GHD in childhood or known pituitary disease. GHD is likely if the patient has documented deficiency of other pituitary hormones including TSH, ACTH, and gonadotropins [1]. In adulthood, GHD can present with reduced energy and depressed mood. GH-deficient subjects have a diminished sense of physical and psychological well-being (e.g., less energy, emotional lability, sense of social isolation, and diminished libido). GH lack contributes to decreased lean body mass and increased abdominal adiposity. Exercise capacity is impaired and there is decreased muscle mass and muscle strength. The contractile properties of muscles are affected in patients with GHD [24]. Fracture risk is increased since bone density, particularly in the lumbar spine, is reduced in some patients with adult-onset GHD. Levels of serum low-density-lipoprotein (LDL) cholesterol may be increased, whereas high-density-lipoprotein (HDL) cholesterol remains normal. Deficiency of GH may contribute to the increased cardiovascular mortality found in patients with hypopituitarism who receive replacement hormones other than GH.
Biochemical Diagnosis
Since GH is secreted in a pulsatile manner, a random GH level is not helpful in the diagnosis of GHD. GH values in a normal individual may vary from undetectable (during an interpulse interval) to more than 40 mg/l. GH secretion is affected by food ingestion; it is suppressed by hyperglycemia and stimulated by amino acids and hypoglycemia. Slow-wave sleep is also associated with increased GH secretion. Insulin-like growth factor-1 (IGF-1, previously termed somatomedin C) is a more reliable screening test for GHD. The synthesis of IGF-1 is GH dependent. It is easier to interpret the age- and gender-specific IGF-1 level given its longer half-life and more steady blood level. A serum IGF-1 concentration below the age- and gender-specific lower limit of normal in a patient who has pituitary disease supports the diagnosis of GHD [2].
If one suspects GHD, a stimulation test is required. Provocative tests of GH secretion include insulin-induced hypoglycemia and arginine (ARG) combined with levodopa (L-DOPA) [25, 26]. Of these, insulin-induced hypoglycemia is most commonly used and has been described earlier in this section.
In the ARG/L-DOPA test, L-DOPA 500 mg PO is given at the start of the infusion of ARG 30 g in a 10% solution over 30 minutes. Blood is obtained 0, 30, 60, 90, and 120 minutes for GH, and a serum GH less than or equal to 1.5 μg/l provides 95% sensitivity and 79% specificity for the diagnosis of GHD. A lowered target GH 0.25 μg/l improves specificity to 95% but lowers sensitivity to 75%. Weaker stimulation tests include ARG, clonidine, or L-DOPA alone. All tests of GH secretion are more likely to give false-positive results in obesity. All the tests are performed in the morning after an overnight fast.
Treatment
GH is most often given as a daily subcutaneous injection at bedtime. The recommended starting dose for adults is 0.006 mg/kg/d with a maximal dose of 0.0125 mg/kg/d. Dosage adjustment is based on the clinical response and the normalization of IGF-1 matched for age and gender. Use of GH replacement is not associated with an obvious early increase in the rate of hypothalamic or pituitary tumor recurrence if pituitary radiation is given postoperatively. GH replacement increases lean body mass, decreases fat mass (particularly abdominal fat), increases bone density, and increases serum HDL levels. Improvement in exercise tolerance, muscle strength, psychosocial assessments, and mortality related to cardiovascular disease has been observed in adults with hypopituitarism that has been treated with GH [24]. An open clinical trial assessing carotid artery intimal wall thickness (IWT) in GH-deficient adults demonstrated a potent
inhibitory effect of 1-year GH replacement on IWT progression, which was maintained after 2 years [27]. The rapid effect of GH replacement on IWT may indicate a beneficial effect of GH treatment on the vascular system. The most common side effects of GH include fluid retention, carpal tunnel syndrome, and arthralgia; these side effects are usually dose related and disappear with dose reduction [28]. The contraindications for GH use include malignancy and diabetic retinopathy.
inhibitory effect of 1-year GH replacement on IWT progression, which was maintained after 2 years [27]. The rapid effect of GH replacement on IWT may indicate a beneficial effect of GH treatment on the vascular system. The most common side effects of GH include fluid retention, carpal tunnel syndrome, and arthralgia; these side effects are usually dose related and disappear with dose reduction [28]. The contraindications for GH use include malignancy and diabetic retinopathy.
Prolactin Deficiency
PRL is the only pituitary hormone that is under tonic inhibition (via dopamine); PRL deficiency is rare and it tends to occur in conjunction with other PHDs. The main clinical presentation of PRL deficiency is inability to lactate in women.
Treatment
Hormone replacement is possible for all the target organ hormones (e.g., cortisol, thyroxine, estrogen, and testosterone) and some of the pituitary hormones (e.g., gonadotropins, GH). Replacement must be tailored to the individual hormone deficiency and, if possible, it should not be instituted until the hypothalamic— pituitary—target organ axis has been assessed. For example, thyroid hormone replacement before institution of glucocorticoid therapy in a patient with cortisol deficiency may precipitate adrenal crisis.
Prolactin
Measurement of a random serum PRL level is useful if the level is unmeasurable or markedly elevated. An unmeasurable level may suggest hypopituitarism. Although PRL concentrations vary during the day, being lower in the afternoon, the time of sampling is usually not critical.
Pituitary Hormone Excess
Thyrotropin Excess
Clinical Presentation
TSH-secreting tumors comprise less than 2% of all pituitary tumors, and most patients have the typical symptoms and signs of hyperthyroidism with diffuse goiter in 95% and visual field defects in 40% [30]. About 25% of patients have cosecretion of other pituitary hormones, usually either PRL or GH. Because of this cosecretion, patients may exhibit galactorrhea and/or the features of acromegaly in addition to hyperthyroidism.
Biochemical Diagnosis
The characteristic thyroid function tests in these patients are normal or high serum TSH with elevated free T3 and free T4 concentrations. Up to 30% of patients will have TSH values within the normal range. It is important to remember that the differential diagnosis of high circulating thyroid hormones with nonsuppressed TSH includes syndromes of resistance to thyroid hormone as well as interferences in either or both the TSH and thyroid hormone assays. The most common cause of the combination of high thyroid hormones and nonsuppressed TSH was interference in the free T4 assay. To determine if this is the case, a free T4 by equilibrium dialysis should be obtained. If the result is normal, then assay interference is the likely cause. If not normal, then the much less common conditions of TSH-producing pituitary tumor (TSHoma) or thyroid hormone resistance need to be considered, and the two must be distinguished. Compared to patients with thyroid hormone resistance, those with TSHomas are more likely to be clinically hyperthyroid, have increased levels of α-subunit (TSH is a heterodimer comprised of a common α-subunit and a unique β-subunit),
have high sex hormone—binding globulin (a thyroid hormone—dependent protein and thus a measure of the tissue effect of thyroid hormone), and have a pituitary lesion identified by sellar MRI. Furthermore, exogenous administration of T3 (80-100 μg/d for 8-10 days) may suppress TSH in those with hormone resistance but not in those with TSHomas. This test is contraindicated in elderly patients or in those with coronary disease. People with TSHomas do not have a TSH response to TRH, another distinguishing feature from hormoneresistant states. However, TRH is no longer available for general clinical use. Since the hormone resistance syndromes are inherited, there may be a family history.
have high sex hormone—binding globulin (a thyroid hormone—dependent protein and thus a measure of the tissue effect of thyroid hormone), and have a pituitary lesion identified by sellar MRI. Furthermore, exogenous administration of T3 (80-100 μg/d for 8-10 days) may suppress TSH in those with hormone resistance but not in those with TSHomas. This test is contraindicated in elderly patients or in those with coronary disease. People with TSHomas do not have a TSH response to TRH, another distinguishing feature from hormoneresistant states. However, TRH is no longer available for general clinical use. Since the hormone resistance syndromes are inherited, there may be a family history.
Treatment
The primary treatment for TSHomas is surgery. If surgery is contraindicated, radiotherapy can be considered. However, experience with somatostatin analogs (SSAs) or dopamine receptor agonist is promising since many TSH-producing pituitary tumors have somatostatin and/or dopamine receptors. After therapy, there may be transient or permanent CH [29].
Nonfunctional Adenomas and Gonadotropin-Producing Adenomas
Overview
Nonfunctional pituitary adenomas (NFPAs) were classically subdivided as gonadotropin-producing adenomas, null cell adenomas, and oncocytomas based on structural and immunohistochemical characteristics [30]. Gonadotropin-producing adenomas stain positively for FSH, LH, and/or their subunits (LH and FSH are heterodimers comprised of a common α-subunit and a unique β-subunit), whereas null cell adenomas and oncocytomas have negative hormonal staining. Gonadotropin-producing adenomas, null cell, and oncocytomas account for about 85% of operated nonfunctioning pituitary adenomas. The remaining 15% are silent adenomas, which express hormones as detected by immunocytochemistry but do not secrete them and thus do not lead to hormone-related clinical symptoms. Most oncocytomas and null cell adenomas belong to the gonadotropin-producing adenoma family, since they express steroidogenic factor 1 (SF-1), a transcription factor whose pituitary expression is specific to the gonadotrope cell lineage. So, despite the negative staining for gonadotropins, these tumors have been shown to secrete LH, FSH, and a-subunits in vitro.
Clinical Presentation and Biochemical Diagnosis
Gonadotropin-producing adenomas usually come to clinical attention as macroadenomas when they cause neurologic symptoms. They may also be found on imaging for an unrelated reason. Impaired vision is the most common presenting symptom, caused by suprasellar extension of the adenoma compressing the optic chiasm. Other presenting neurologic symptoms include headache, diplopia, cerebrospinal fluid rhinorrhea, and pituitary apoplexy induced by sudden hemorrhage. Importantly, apoplexy can be precipitated by administration of a drug that stimulates gonadotrope adenomas, such as GnRH analog treatment of prostate cancer.
Hormonal deficiencies can also be the initial presentation. These are due to compression of nonadenomatous pituitary cells by the macroadenoma and by mechanical compression of portal vessels and pituitary stalk. This stalk compression leads to diminished delivery of hypothalamic hormones and dopamine to pituitary cells. The decrease of hypothalamic hormones may lead to deficiencies of most of the anterior pituitary hormones. In contrast, the decreased delivery of dopamine, an inhibitor of PRL secretion, causes an increase in circulating
PRL. This increased PRL in the context of a non-PRL-secreting pituitary tumor is termed pseudoprolactinoma. The diagnosis is established by finding abnormalities of the subunits of LH and FSH that are characteristic of a gonadotrope adenoma and follow-up imaging by sellar MRI.
PRL. This increased PRL in the context of a non-PRL-secreting pituitary tumor is termed pseudoprolactinoma. The diagnosis is established by finding abnormalities of the subunits of LH and FSH that are characteristic of a gonadotrope adenoma and follow-up imaging by sellar MRI.
The least common clinical presentation is one that results from hormonal abnormalities, as gonadotrope adenomas are hormonally inefficient and the LH and FSH produced are usually not sufficient to result in hormonal derangements. Also, only about 35% of these tumors secrete enough gonadotropins to raise the serum levels [31], and the hormone combinations differ in men and women. In men, an elevated basal serum FSH concentration with an intrasellar mass lesion usually indicates that the lesion is a gonadotrope adenoma. The diagnosis is strengthened if the patient also has a supranormal level of the free α-subunit and serum LH accompanied by an elevated testosterone level. In women, recognizing the gonadotrope origin of an intrasellar mass on the basis of serum intact FSH and LH is more difficult, especially in menopausal woman. However, a gonadotrope adenoma is suspected if there is a markedly elevated FSH level associated with a subnormal LH concentration, as this pattern is not likely in the postmenopausal state or in premature ovarian failure. An elevated serum free α-subunit concentration is also helpful. An elevated serum estradiol with an FSH concentration that is not suppressed associated with endometrial hyperplasia and polycystic ovaries by ultrasound in a woman of premenopausal age who presents with amenorrhea or oligomenorrhea strongly suggests a gonadotrope adenoma—secreting FSH and causing ovarian hyperstimulation. It is important to remember that a “normal” FSH is not appropriate when the estradiol is markedly elevated and the LH suppressed. Prepubertal girls may present with breast development, vaginal bleeding, and abdominal distension and young boys with premature puberty.
Treatment
Because of the slow growth of microadenomas (see section on Pituitary Incidentalomas), small nonfunctional or gonadotropin-secreting lesions can be treated conservatively and followed with serial imaging. Adenoma enlargement with mass effect symptoms is an indication for more aggressive management with surgery and or irradiation. However, if the tumor presents as a macroadenoma as it often does, more aggressive treatment can be considered earlier. The use of dopamine receptor agonists and SSAs is helpful in shrinking or at least preventing further growth of some of these tumors and can be used in conjunction with surgery and irradiation [31].
Prolactin Excess
Hyperprolactinemia is described in a subsequent section of this chapter
Pituitary Incidentalomas
Pituitary adenomas detected as incidental findings on head MRI or CT scans are called pituitary incidentalomas. Patients with pituitary incidentalomas should be screened for hypersecretion of prolactin (PRL), acromegaly (IGF-1), CD (salivary and urine cortisol levels), and TSH, and those with macroadenomas should also be screened for hypopituitarism and for visual field defects if the tumor abuts the optic chiasm. Growth of nonfunctioning pituitary adenomas without treatment occurs in about 10% of microadenomas and 24% of macroadenomas over 10 years. In the absence of hypersecretion, hypopituitarism, or visual field defects, patients may be followed up by periodic screening by MRI. Growth of a pituitary incidentaloma is an indication for surgery.
IMAGING OF THE HYPOTHALAMIC—PITUITARY SYSTEM
Radiologic Imaging Techniques
MRI specifically focused on the sellar region is the imaging modality of choice for visualization of hypothalamic—pituitary anatomy. Standard protocol consists of 2 to 3 mm sections of sagittal and coronal T1- and T2-weighted images with and without contrast enhancement. This technique delineates the pituitary gland, stalk, optic tracts, and surrounding structures such as the cavernous sinus. The more widely spaced cuts of a routine brain MRI are often inadequate to visualize small pituitary tumors. The anterior pituitary is isodense with white matter, and the posterior pituitary exhibits a bright spot of high signal intensity thought to reflect the presence of vasopressin localized within neurosecretory vesicles. Lack of this feature can represent a nonspecific indicator of central diabetes insipidus (CDI). The size of the pituitary gland varies with age and sex. On average, it measures between 3 and 8 mm in height but is generally larger in females than males. The pituitary gland can transiently enlarge during puberty, pregnancy, and in the postpartum period. The stalk should not exceed 4 mm in diameter; a thickened stalk can indicate hypophysitis, histiocytosis, lymphoma, or granulomatous disease [32, 33].
Most microadenomas (tumors ≤10 mm) are hypointense on T1-weighted magnetic resonance (MR) images in relation to adjacent pituitary tissue. This difference is accentuated after contrast enhancement with gadolinium (Fig. 1a). Microadenomas can cause gland asymmetry or stalk displacement. Macroadenomas (>10 mm), which tend to be more vascular tumors, show intermediate signal on unenhanced T1-weighted images and can enhance after gadolinium administration. They often enlarge the sella turcica by gradual remodeling of the bony fossa and show frequent extrasellar extension [32]. The normal pituitary gland can be displaced superiorly, although in 40% of cases, the pituitary gland may not be detectable [34]. These tumors can also grow upward toward the optic chiasm and cause draping of the nerves over the tumor, which is often accompanied by visual field abnormalities (Fig. 1b). Subacute hemorrhage may occur, demonstrating high signal on T1- and T2-weighted images due to the presence of hemoglobin degradation products.
CT is superior to MRI in demonstrating cortical bone, often critical in the case of pituitary adenomas causing erosion of the sellar floor and clinoid bones. CT can also identify intralesional calcifications characteristic of craniopharyngiomas, meningiomas, and chordomas [35].
The radiolabeled SSA111In-pentetreotide may be used to detect the presence of tissues expressing somatostatin receptors [35]. Octreotide scanning may be used to detect both pituitary and ectopic neuroendocrine tumors expressing these somatostatin receptors. It provides both a visual image and a physiologic measure, and increased uptake may predict the response of medical therapy with SSAs. However, given that these receptors are expressed by normal endocrine tissue as well as nonfunctioning adenomas, the specificity of this procedure is limited. It can be helpful when coupled with an abnormal CT or MRI result to confirm diagnosis.
Positron emission tomography (PET) has an evolving role in the evaluation of sellar masses. The use of18F-fluoroethyl-spiperone (FESP) PET has shown to be specific for differentiating adenomas from other sellar tumors such as craniopharyngiomas and meningiomas [36]. The uptake of FESP is also consistently higher in nonfunctioning adenomas [36]. Currently, the use of PET imaging in the assessment of pituitary tumors is at the research level.
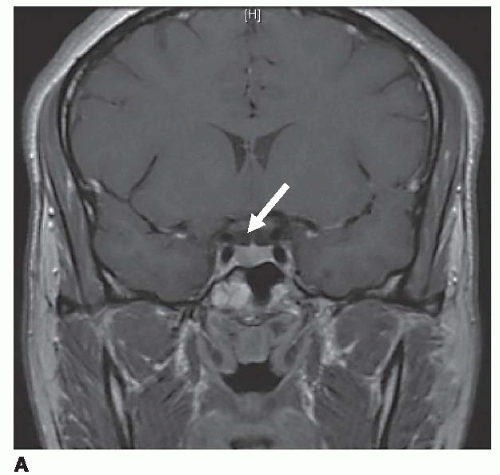 Figure 1a. Coronal T1-weighted MR image through pituitary fossa showing a pituitary microadenoma with hypo-enhancement of the right pituitary gland (arrow). |
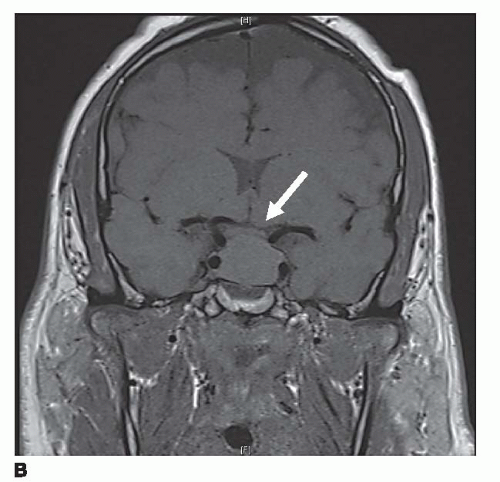 Figure 1b. Coronal T1-weighted MR image through pituitary fossa showing a pituitary microadenoma elevating and compressing optic chiasm (arrow). |
Other Sellar Lesions
Certain pituitary lesions have specific signal characteristics, which may aid in diagnosis. Rathke cleft cysts and craniopharyngiomas have a common origin from remnants of the Rathke pouch. Craniopharyngiomas are benign yet aggressive squamous epithelial tumors that are predominantly suprasellar. Prominent cystic portions with wall enhancement or varying signal intensities of mixed cystic and solid components with calcification are typical imaging features [32]. CT aids in identification of calcifications, which represent the hallmark of craniopharyngiomas. Rathke cleft cysts are benign lesions containing mucoid material and usually show a sellar epicenter, smooth contour, and reduced MR signal intensity on T1- and increased on T2-weighted imaging [34].
Meningiomas, primitive germ cell tumors, chordomas, and metastases also represent neoplasms, which appear in the sellar region. Meningiomas may arise from any dural site. Characteristic features include dense homogenous enhancement after gadolinium, dural tail enhancement, and adjacent hyperostosis (bone thickening) [34]. Germinomas are generally isointense on T1- and T2-weighted imaging. Chordomas, arising from the notochordal remnants of the clivus, typically produce bony destruction and can lead to cranial neuropathy and neurologic symptoms. Pituitary metastases are found in 1% to 2% of patients, with breast and lung carcinoma being the most common primary sources. Interestingly, the posterior pituitary can be involved in 70% of these cases, and patients may present with DI. The MRI/CT features are those of an enhancing mass, but potential multiplicity of lesions, with a history of a known primary tumor, often leads to the diagnosis.
DISORDERS OF THE PROLACTIN SYSTEM
Pathophysiology
PRL synthesis and secretion by pituitary lactotroph cells is tonically suppressed by hypothalamic dopamine, which reaches the lactotrophs through the portal venous system. PRL acts to induce and maintain lactation in the primed breast. Factors stimulating PRL synthesis and secretion include estrogen, TRH, epidermal growth factor, and dopamine receptor antagonists. Hyperprolactinemia is the most common neuroendocrine condition in clinical practice. Lactotroph adenomas account for approximately 40% of all pituitary adenomas, with 90% being microadenomas [37]. Hyperprolactinemia may also develop due to either impairment of the hypothalamic production of dopamine or compression of the pituitary stalk that impairs dopamine transport to the pituitary. Medications that cause elevated PRL levels include antidepressants and antipsychotics, as well as some antihypertensive agents, opiates, and H2-receptor blockers [38]. PRL levels can also be increased in patients with chronic renal insufficiency or hepatic failure, usually due to reduced clearance. The many etiologies of hyperprolactinemia are listed in Table 1.2.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


