Introduction
Hypothalamic stimulatory and inhibitory hormones traverse the hypophyseal-portal circulation to target specific cells of the pituitary gland. The pituitary cells synthesize and secrete trophic hormones, which target organs to secrete hormones, which then affect peripheral tissues ( Figs. 20.1 and 20.2 ). Hypopituitarism, also known as pituitary insufficiency, refers to one or more pituitary deficiencies caused by pituitary or hypothalamic disease.
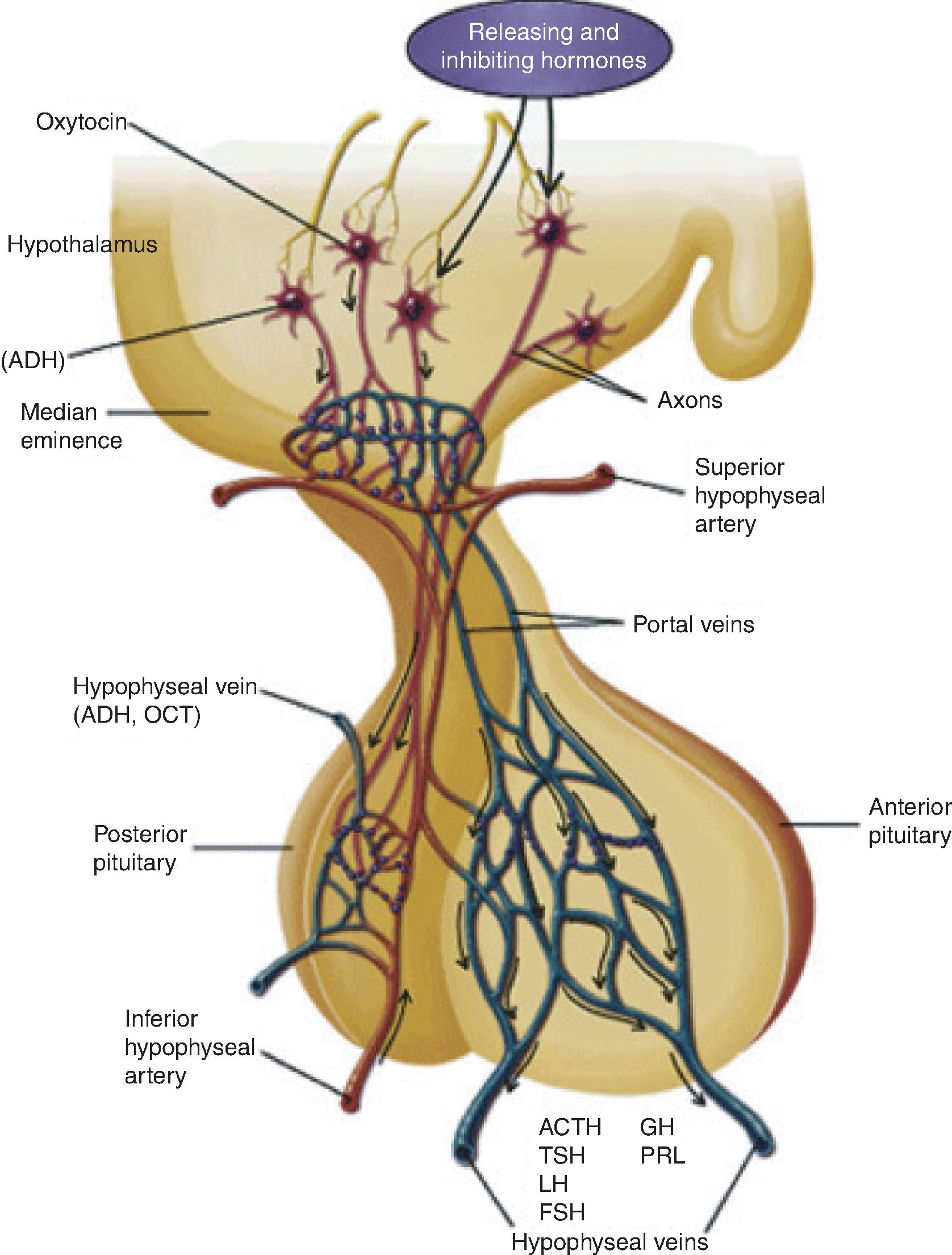
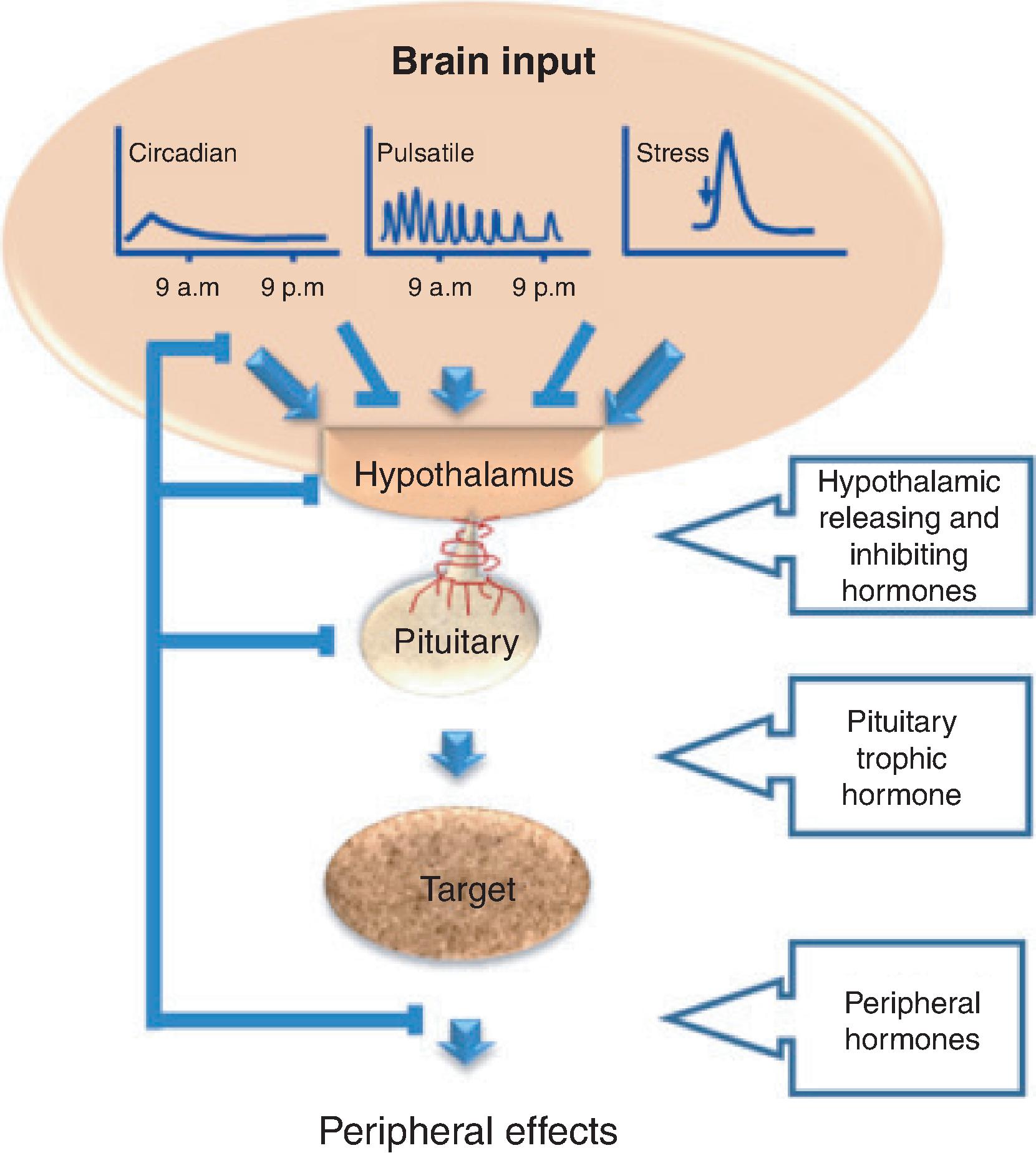
Hypopituitarism can be congenital or acquired and transient or permanent. The clinical manifestations may vary depending on the severity of the hormone deficit. In most scenarios, the secretion of gonadotropins and growth hormone (GH) is more likely to be affected than adrenocorticotropic hormone (ACTH) and thyroid-stimulating hormone (TSH), but isolated deficiencies of any hormone can occur. Posterior pituitary insufficiency, characterized by decreased circulating antidiuretic hormone (ADH), leading to central diabetes insipidus (DI), will be discussed in Chapter 19.
EPIDEMIOLOGY
The prevalence of hypopituitarism is approximately 450 cases per million with an incidence of about 40 new cases per million per year. , There is high mortality associated with hypopituitarism.
ETIOLOGIES
Hypopituitarism can be congenital or acquired and can arise from structural or functional disruption of the hypothalamus, portal system, or pituitary gland ( Table 20.1 ). Patients with hypothalamic or pituitary masses, developmental craniofacial abnormalities, inflammatory disorders, neuro-granulomatous disease, head trauma, prior brain irradiation, or prior skull base surgery, and those who have previously experienced pregnancy-associated hemorrhage, should be screened for hypopituitarism.
| Neoplastic |
| Pituitary adenoma or carcinoma |
| Metastatic disease |
| Craniopharyngioma |
| Lymphoma |
| Parasellar masses (meningioma, germinoma, glioma) |
| Traumatic |
| Surgery |
| Radiation |
| Traumatic brain injury |
| Vascular |
| Sheehan’s syndrome |
| Apoplexy |
| Carotid aneurysm |
| Subarachnoid hemorrhage |
| Infiltrative/Inflammatory |
| Hypophysitis |
| Sarcoidosis |
| Wegner granulomatosis |
| Histiocytosis X |
| Hemochromatosis |
| Infectious |
| Bacterial infections |
| Fungal infections |
| Parasitic infections |
| Congenital |
| Mutations of PROP1, HESX1, LHX3, LHX4, POU1F1, POMC, TPIT |
| Drug-Induced |
| Glucocorticoids |
| Opiates |
| Bexarotine |
DIAGNOSIS
The diagnosis of hypopituitarism relies on the measurement of basal and stimulated secretion of anterior pituitary hormones and the hormones secreted by the target glands. Magnetic resonance imaging (MRI) of the hypothalamus and pituitary with and without gadolinium contrast is the imaging test of choice. Genetic testing may be indicated in congenital or syndromic disease.
Central Adrenal Insufficiency
INTRODUCTION
Hypothalamic corticotropin-releasing hormone (CRH) stimulates ACTH secretion by pituitary corticotroph cells. ACTH stimulates cortisol synthesis and secretion from the adrenal gland. ACTH is secreted in a pulsatile manner and follows a circadian rhythm, reaching peak levels in the early morning and nadirs around midnight. Cortisol exerts negative feedback inhibition on pituitary ACTH and hypothalamic CRH secretion ( Fig. 20.3 ). Central adrenal insufficiency (AI) occurs when there is loss of hypothalamic CRH (tertiary adrenal insufficiency) or pituitary release of ACTH (secondary adrenal insufficiency). The most common cause of central AI is iatrogenic due to suppression of the hypothalamic-pituitary-adrenal (HPA) axis after glucocorticoid administration.
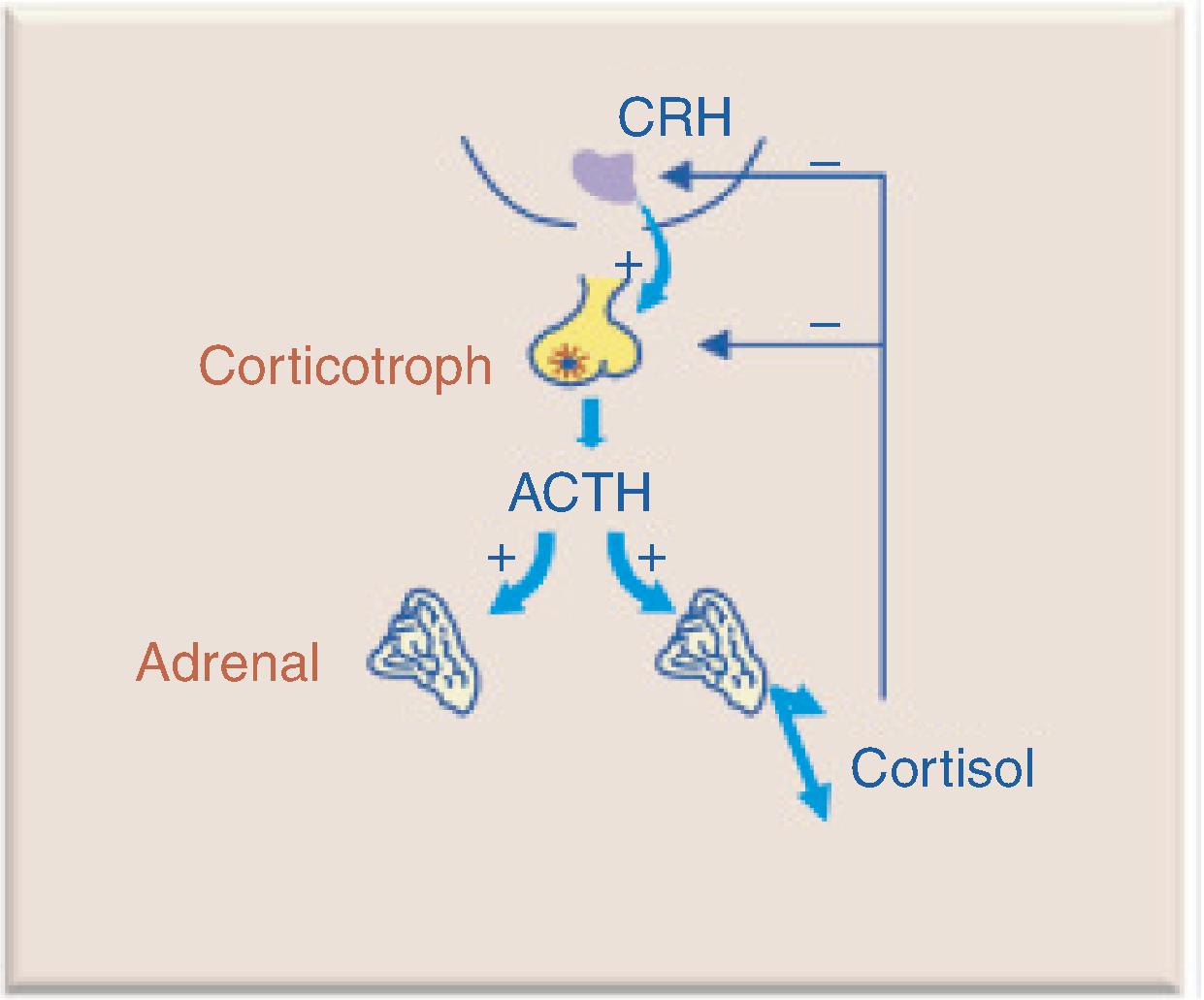
Acute adrenal insufficiency is a life-threatening emergency presenting with hypotension or hypovolemic shock. It can occur abruptly (e.g., pituitary apoplexy) or insidiously during times of physical stress in previously undiagnosed patients or those with known AI who fail to properly increase their replacement doses.
CLINICAL PRESENTATION
Central AI results in decreased secretion of cortisol and adrenal androgens. It can present with lethargy, weakness, nausea, vomiting, and hypotension. Signs of mineralocorticoid deficiency (salt craving, hyperkalemia) are not present, as adrenal mineralocorticoid secretion is preserved. Hyponatremia can occur secondary to inappropriate secretion of ADH resulting from glucocorticoid deficiency ( Box 20.1 ).
DIAGNOSTIC EVALUATION
We are unable to routinely measure CRH, and ACTH measurements are not very reliable because of the short plasma half-life, pulsatile secretion, and rigorous specimen handling requirements. Therefore we rely on cortisol testing for the diagnosis of adrenal insufficiency. However, due to the circadian changes in cortisol, a random cortisol level is not helpful. The first-line test for diagnosing central AI is measuring serum cortisol levels at 8:00 to 9:00 a.m. A cortisol level less than 3 g/dL is indicative of AI and a cortisol level greater than 15 g/dL likely excludes an AI diagnosis. For morning cortisol levels between 3 and 15 g/dL, stimulation testing is recommended. Central AI can be diagnosed using insulin tolerance and the low-dose and standard-dose ACTH stimulation tests. The standard-dose test using 250 μg cosyntropin (synthetic ACTH 1-24) administered intravenously or intramuscularly is recommended. Peak cortisol levels less than 18.1 g/dL at 30 or 60 minutes indicate AI. With the use of new cortisol assays using more specific monoclonal antibody immunoassays or liquid chromatography with tandem mass spectrometry (LC-MS/MS), a lower cut-off for a normal response to ACTH testing of greater than 14.5 μg/dL has been proposed. Furthermore, cortisol cut-offs for provocative testing are based on total cortisol levels and assume normal cortisol-binding globulin (CBG). Albumin is often used as a surrogate for CBG and lower cut-offs for normal cortisol response to ACTH testing or use of free cortisol levels may be considered in patients with low albumin. Exogenous glucocorticoids should be withheld 18 to 24 hours prior to testing ( Table 20.2 ).
| Test | Procedure | Interpretation | Comments |
|---|---|---|---|
| Serum cortisol | Morning (8:00–9:00 a.m.) basal testing | Normal: serum cortisol level >15 μg/dL | Assumes normal CBG |
| Insufficient: <3 μg/dL | |||
| Indeterminate: 3–15 μg/dL | |||
| Insulin tolerance test | Administer insulin (0.05–0.15 U/kg IV)Serum measurements of cortisol and glucose at 0, 30, 60 min | Blood glucose <40 mg/dL required for interpretationNormal: peak cortisol >18 μg/dLInsufficient: serum cortisol level <18 μg/dL | Symptomatic hypoglycemia contraindicated in elderly patients, those with seizure disorders, and cardiac disease |
| Cortisol cut-off may be lower depending on assay | |||
| ACTH stimulation test (standard dose) | Administer synthetic ACTH 1-24 250 μg IV or IMSerum measurements of cortisol at 0, 30, 60 min | Normal: serum cortisol level <18 μg/dLInsufficient: serum cortisol level <18 μg/dL | Cortisol cut-off may be lower depending on assayFalse negative results in acute central AI |
MANAGEMENT
Central AI can lead to adrenal crisis, which is a life-threating emergency. Adrenal crisis occurs when the HPA axis cannot produce sufficient cortisol in response to an increased need ( Box 20.2 ). Patients should be treated with aggressive hydration and stress-doses of glucocorticoids. For suspected adrenal crisis due to secondary AI, treat with an immediate parenteral injection of 50 to 100 mg hydrocortisone followed by 200 mg/day for the next 24 hours. The dose should be reduced to hydrocortisone 100 mg/day in divided doses the following day. If the patient is improving, and without illness or planned-for invasive procedures, the goal should be to taper down to physiologic doses of glucocorticoids within 3 days.
- ■
Acute stress in patient with known central AI
- ■
Pituitary infarction/apoplexy
- ■
After surgical cure of Cushing’s syndrome
- ■
Abrupt withdrawal of exogenous glucocorticoids
For long-term treatment of central AI, hormonal replacement should try to mimic the physiologic cortisol pattern. Hydrocortisone 15 to 20 mg total daily dose should be given in divided doses two to three times a day. Patients should take the highest dose in the morning upon awakening, and the second in the afternoon (two-dose regimen) or the second and third at lunch and late afternoon, respectively (three-dose regimen). Longer-acting glucocorticoids such as prednisone or prednisolone 2.5 to 7.5 mg/day or dexamethasone 0.25 to 0.75 mg/day may be used in selected patients for improved compliance or convenience ( Table 20.3 ). Retrospective studies of patients taking higher doses and longer-acting glucocorticoids show a tendency toward adverse metabolic consequences including weight gain, dyslipidemia, and type 2 diabetes mellitus. Slow-release preparations of hydrocortisone and hydrocortisone infusion pumps are in clinical development. Glucocorticoid dosing should be adjusted based on clinical symptoms, side effects, and comorbidities.
| Formulation | Dosing | Comments |
|---|---|---|
| Hydrocortisone | 15–20 mg/day | Two or three times daily |
| Potential for small dose adjustments | ||
| Prednisolone/Prednisone | 2.5–7.5 mg/day | Long-acting physiologic effect |
| Risk of overtreatment | ||
| Dexamethasone | 0.25–0.75 mg/day | Long-acting physiologic effect |
| Variable metabolism | ||
| Risk of overtreatment |
Because AI may mask the presence of partial DI, patients should be monitored for the development of DI after starting glucocorticoid replacement.
Once diagnosed with central AI, patient education is considered the key to preventing adrenal crisis. Patients should receive detailed information on their disease and learn how to self-adjust glucocorticoids during times of stress or illness ( Table 20.4 ). Patients should carry an emergency card, bracelet, or necklace, and injectable glucocorticoids for emergency administration.
| Scenario | Change to Maintenance Dose |
|---|---|
| Febrile illness | Double or triple dose for 3 days |
| Minor trauma | Double or triple dose for 2–3 days |
| Minor surgical procedures | Double or triple dose for the day of surgery |
| Moderate surgical procedures | Doses equivalent to hydrocortisone 50–75 mg in divided doses for the day of surgery and the first postoperative day, with a return to the usual dose on the second postoperative day |
| Major surgical procedures | Doses equivalent to hydrocortisone 100–150 mg in divided doses for the day of surgery and the next 2–3 days, then return to the usual dose |
| Critical illness | Doses equivalent to hydrocortisone 200–300 mg/day in divided doses for 3–7 days, with tapered withdrawal guided by clinical response |
| Major trauma | Doses equivalent to hydrocortisone 100–150 mg in divided doses for the day of trauma and the next 2–3 days, then return to the usual dose |
Central Hypothyroidism
INTRODUCTION
Hypothalamic thyrotropin-releasing hormone (TRH) stimulates TSH secretion by pituitary thyrotroph cells. TSH binds to receptors on the thyroid gland and activates adenylyl cyclases, stimulates iodine uptake, and leads to the synthesis and secretion of thyroid hormones thyroxine (T4) and triiodothyroinine (T3). T4 and T3 exert negative feedback inhibition on pituitary TSH and hypothalamic TRH secretion ( Fig. 20.4 ). Central hypothyroidism occurs when there is loss of hypothalamic TRH (tertiary hypothyroidism) or pituitary production and release of TSH (secondary hypothyroidism).
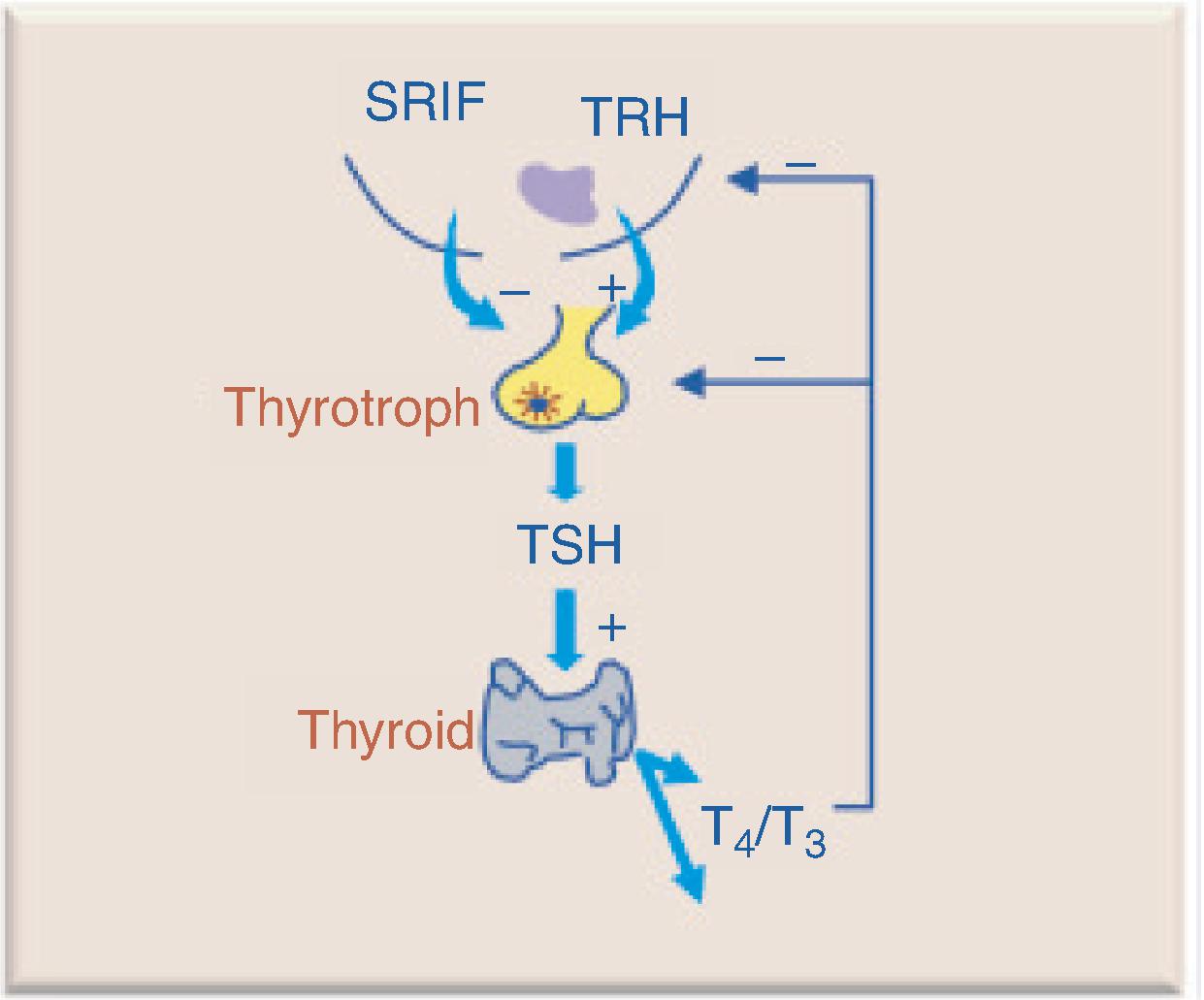
CLINICAL PRESENTATION
Central hypothyroidism can present with lethargy, constipation, cold intolerance, bradycardia, weight gain, and hyponatremia. The presentation is variable, with some patients being asymptomatic and others having profound symptoms of hypothyroidism ( Box 20.3 ).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


