Abbreviations
- ACTH Adrenocorticotrophic hormone
- ACE Angiotensin-converting enzyme
- ADP Adenosine diphosphate
- ALS Acid-labile subunit
- ATP Adenosine triphosphate
- GDH Glutamate dehydrogenase
- GIP Glucose-dependent insulinotropic polypeptide
- GLP-1 Glucagon-like peptide 1
- G-6-Pase Glucose-6-phosphatase
- HbA1c Hemoglobin A1c
- ICMA Immunochemiluminometric
- IGF-II Insulin-like growth factor II
- IGFBP3 IGF-binding protein-3
- Kir Inward rectifier potassium channel
- NADH Nicotinamide adenine dinucleotide (reduced form)
- NICTH Nonislet cell tumor hypoglycemia
- NIPHS Noninsulinoma pancreatogenous hypoglycemic syndrome
- PEPCK Phosphoenolpyruvate carboxykinase
- RIA Radioimmunoassay
- SSTR Somatostatin receptor
- SUR Sulfonylurea receptor
Hypoglycemic Disorders: Introduction
Circulating plasma glucose concentrations are kept within a relatively narrow range by a complex system of interrelated neural, humoral, and cellular controls. Under the usual metabolic conditions, the central nervous system is wholly dependent on plasma glucose and counteracts declining blood glucose concentrations with a carefully programmed response. This is often associated with a sensation of hunger and, as the brain receives insufficient glucose to meet its metabolic needs (neuroglycopenia), an autonomic response is triggered to mobilize storage depots of glycogen and fat. In the postabsorptive state, hepatic glycogen reserves and gluconeogenesis from the liver and kidney directly supply the central nervous system with glucose, which is carried across the blood-brain barrier by a specific glucose transport system, while the mobilization of fatty acids from triglyceride depots provides energy for the large mass of skeletal and cardiac muscle, renal cortex, liver, and other tissues that utilize fatty acids as their basic fuel, thus sparing glucose for use by the tissues of the central nervous system.
The normal lower limit of fasting plasma glucose is typically 70 mg/dL (3.9 mmol/L). Lower values may occur during prolonged fasting, strenuous exercise, or pregnancy or may occur as a laboratory artifact. In normal men, plasma glucose does not fall below 55 mg/dL (3 mmol/L) during a 72-hour fast. However, for reasons that are not clear, normal women may experience a fall to levels as low as 30 mg/dL (1.7 mmol/L) despite a marked suppression of circulating insulin to less than 5 μU/mL. They remain asymptomatic in spite of this degree of hypoglycemia, presumably because ketogenesis is able to satisfy the energy needs of the central nervous system. Basal plasma glucose declines progressively during normal pregnancy, and hypoglycemic levels may be reached during prolonged fasting. This may be a consequence of a continuous fetal consumption of glucose and diminished availability of the gluconeogenic substrate alanine. The cause of these diminished alanine levels in pregnancy is unclear. The greatly increased glucose consumption by skeletal muscle that occurs during prolonged strenuous exercise may lead to hypoglycemia (blood glucose <45 mg/dL) despite increases in hepatic glucose production. Whether the hypoglycemia in this circumstance contributes to fatigue or other symptoms in distance runners is unknown.
In vitro consumption of glucose by blood cell elements may give rise to laboratory values in the hypoglycemic range. This can be avoided by adding a small amount of the metabolic inhibitor sodium fluoride to collection tubes used for specimens containing increased numbers of blood cells (as in leukemia, leukemoid reactions, or polycythemia) and keep samples on ice until separated.
Pathophysiology of the Counterregulatory Response to Neuroglycopenia
The plasma concentration of glucose that signals the need by the central nervous system to mobilize energy reserves depends on a number of factors, such as the status of blood flow to the brain, the integrity of cerebral tissue, the prevailing arterial level of plasma glucose, the rapidity with which plasma glucose concentration falls, and the availability of alternative metabolic fuels.
A hierarchy of responses has been shown to occur as plasma glucose falls in healthy young volunteers, with hormonal counterregulatory responses being triggered at glucose levels slightly higher (∼67 mg/dL [3.7 mmol/L]) than those that induce symptoms of hypoglycemia (Figure 18–1). The first symptoms to appear in healthy people are mediated by autonomic neurotransmitters and occur at plasma glucose levels below 60 mg/dL (3.3 mmol/L). The symptoms consist of tremor, anxiety, palpitations, and sweating, which result from sympathetic discharge; and hunger, which is a consequence of parasympathetic vagal response. Ganglionic blockade and cervical cord section or sympathectomy—but not adrenalectomy—ameliorates these symptoms, indicating that they are due to the release of autonomic neurotransmitters and not dependent on adrenal hormones. As plasma glucose falls below 50 mg/dL (2.8 mmol/L), cerebral neuroglycopenia ensues, consisting of impaired cognition, along with weakness, lethargy, confusion, incoordination, and blurred vision. If counterregulatory responses are inadequate to reverse this degree of profound hypoglycemia, convulsions or coma may occur. This can result in brain damage or death, particularly in those who have not adapted to repeated episodes of hypoglycemia.
Figure 18–1
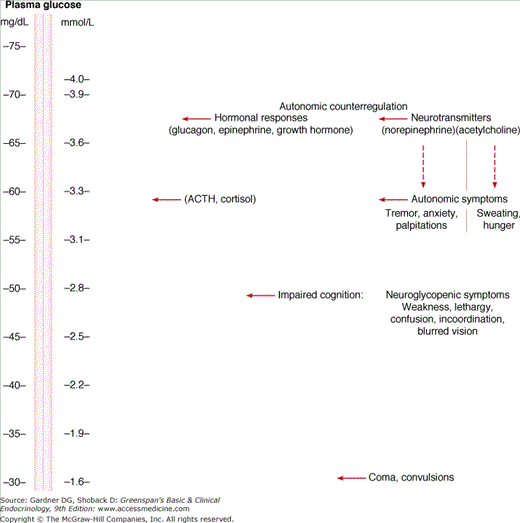
Hierarchy of autonomic responses to progressive stepwise reduction in plasma glucose concentration in healthy volunteers.
(Adapted with permission from Gerich JE, et al. Hypoglycemia unawareness. Endocr Rev. 1991;12:356; and from Service FJ. Hypoglycemia disorders. N Engl J Med. 1995;332:1144.)
In elderly people, however, with compromised cerebral blood supply, neuroglycopenic manifestations may be provoked at slightly higher plasma glucose levels. Patients with chronic hyperglycemia (eg, those with poorly controlled, insulin-treated diabetes mellitus) may experience symptoms of neuroglycopenia at considerably higher plasma glucose concentrations than persons without diabetes. This has been attributed to a downregulated glucose transport system across the blood–brain barrier. Conversely, in patients exposed to chronic hypoglycemia (eg, those with an insulin-secreting tumor or those with diabetes who are receiving excessively tight glycemic control with an insulin pump), adaptation to recurrent hypoglycemia occurs by upregulation of the glucose transporters, which results in hypoglycemic unawareness, whereby they show greater tolerance to hypoglycemia without manifesting symptoms. Restoring and maintaining an adequate supply of glucose for cerebral function proceeds by a series of neurogenic events that act directly to raise the plasma glucose concentration and to stimulate hormonal responses that augment the adrenergic mobilization of energy stores (Table 18–1).
| Alpha-Adrenergic Effects |
| Inhibition of endogenous insulin release |
| Increase in cerebral blood flow (peripheral vasoconstriction) |
| Beta-Adrenergic Effects |
| Hepatic and muscle glycogenolysis |
| Stimulation of plasma glucagon release |
| Lipolysis to raise plasma-free fatty acids |
| Impairment of glucose uptake by muscle tissue |
| Increase in cerebral blood flow (increase in cardiac output) |
| Adrenomedullary Discharge of Catecholamines |
| Augmentation of all of the above alpha- and beta-adrenergic effects |
| Cholinergic Effects |
| Raises level of pancreatic polypeptide |
| Increases motility of stomach |
| Produces hunger |
| Increases sweating |
Insulin: Endogenous insulin secretion is lowered both by reduced glucose stimulation to the pancreatic β cell and by sympathetic nervous system inhibition from a combination of alpha-adrenergic neural effects and increased circulating catecholamine levels. This reactive insulinopenia appears to be essential for glucose recovery, because it facilitates the mobilization of energy from existing energy stores (glycogenolysis and lipolysis); increases hepatic enzymes involved in gluconeogenesis and ketogenesis; increases enzymes of the renal cortex, promoting gluconeogenesis; and at the same time prevents muscle tissue from consuming the blood glucose being released from the liver (Chapter 17).
Catecholamines: Circulating catecholamines—and norepinephrine produced at sympathetic nerve endings—provide muscle tissue with alternative sources of fuel by activating beta-adrenergic receptors, resulting in mobilization of muscle glycogen, and by providing increased plasma-free fatty acids from lipolysis of adipocyte triglyceride. Metabolism of these free fatty acids provides energy to promote gluconeogenesis in the liver and kidney, thereby adding to plasma glucose levels already raised by the glycogenolytic effect of catecholamines on the liver. Their cardiovascular and other side effects provide a signal that diabetic patients learn to recognize as a warning of their need to rapidly ingest absorbable carbohydrate.
Glucagon: Plasma glucagon is released by the beta-adrenergic effects of both sympathetic innervation and circulating catecholamines on pancreatic α cells as well as by the direct stimulation of α cells by the low plasma glucose concentration itself. Data are available suggesting that a falling-off of intra-islet insulin concentration in subjects with functioning pancreatic β cells can release pancreatic α cells from insulin inhibition and thus augment glucagon release during hypoglycemia. This glucagon release increases hepatic output of glucose by direct glycogenolysis as well as by facilitating the activity of gluconeogenic enzymes in the liver but not in the kidney. As shown in Figure 18–2, plasma glucagon appears to be the key counterregulatory hormone affecting recovery from acute hypoglycemia in nondiabetic humans, with the adrenergic-catecholamine response representing a major backup system. However, in most clinical situations, where hypoglycemia develops more gradually, as with inappropriate dosage of insulin or sulfonylureas, or in cases of insulinoma, the role of glucagon may be less influential. When normal volunteers received a prolonged low-dose insulin infusion to produce a gradual decline in plasma glucose levels without waning of insulin levels, the rise of endogenous glucagon contributed much less to counterregulation than after acute hypoglycemia induced by intravenous insulin, which is followed by rapid waning of insulin levels. This finding suggests that glucagon’s role in glucose recovery occurs primarily when the level of insulin wanes.
Figure 18–2
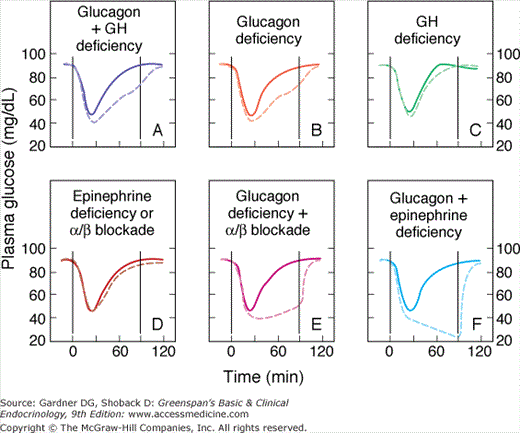
Solid lines show changes in plasma glucose that occur in normal subjects in response to acute intravenous insulin administration. Note the rapid recovery of glucose levels mediated by intact counterregulatory mechanisms. The dashed lines show the response to insulin-induced hypoglycemia in patients with deficiencies of the counterregulatory mechanisms induced as follows: A: Somatostatin infusion (inhibits both glucagon and growth hormone [GH] release). B: Somatostatin infusion plus GH infusion (now with functional isolated glucagon deficiency). C: Somatostatin infusion plus glucagon infusion (isolated GH deficiency). Note return of glucose response to normal, implying that glucagon is the main counterregulatory hormone. D: Bilateral adrenalectomy, leading to epinephrine deficiency, or infusion of phentolamine plus propranolol (alpha and beta blockers, respectively). Note that such deficiencies cause no major abnormality in response to induced hypoglycemia when glucagon is present. E and F: Sympathetic modulation (by phentolamine plus propranolol in E and by bilateral adrenalectomy in F), which seriously impairs the ability to respond to hypoglycemia in the patient made glucagon-deficient by somatostatin infusion.
(Reproduced, with permission, from Cryer PE. Glucose counterregulation in man. Diabetes. 1981;30:261.)
Cortisol: Pituitary corticotrophin (ACTH) is released in association with the sympathetic nervous system stimulation by neuroglycopenia. This results in elevation of plasma cortisol levels, which in turn permissively facilitates lipolysis and actively promotes protein catabolism and conversion of amino acids to glucose by the liver and kidney.
Growth hormone: Pituitary growth hormone is also released in response to falling plasma glucose levels. Its role in counteracting hypoglycemia is less well defined, but it is known to antagonize the action of insulin on glucose utilization in muscle cells and to directly activate lipolysis by adipocytes. This increased lipolysis provides fatty acid substrate to the liver and renal cortex which facilitates gluconeogenesis.
Cholinergic neurotransmitters: Acetylcholine is released at parasympathetic nerve endings, and its vagal effects induce the sensation of hunger that signals the need for food to counteract the hypoglycemia. In addition, postsynaptic fibers of the sympathetic nervous system that innervate the sweat glands to signal hypoglycemia also release acetylcholine—in contrast to all other sympathetic postsynaptic fibers, which without exception release norepinephrine.
Glucose absorption from the gastrointestinal tract ceases by 4 to 6 hours after a meal. During the postabsorptive state which immediately follows, glucose must be produced endogenously from previously stored nutrients to meet the requirements of the central nervous system and other glucose-dependent tissues. These include 125 mg/min of glucose required by the brain and spinal cord as well as an additional 25 mg/min by red blood cells and the renal medulla. It was previously thought that the liver is the only organ involved in glucose production during an overnight fast, but recent data indicate that the renal cortex also has the requisite enzymes for production and release of glucose.
Liver: The liver initially provides glucose by the breakdown of stored hepatic glycogen. However, because these reserves are limited to 80 to 100 g, they begin to be depleted several hours into the postabsorptive state. Thereafter, hepatic glucose production is augmented by gluconeogenesis—the formation of glucose from amino acids, lactate, and glycerol. These substrates are delivered to the liver and kidney from peripheral stores. Muscle and other structural tissues supply amino acids, mainly alanine; blood cell elements supply lactate, the end product of glycolytic metabolism; and adipose tissue supplies glycerol from lipolysis of triglyceride. In addition, oxidation of the free fatty acids released from adipose cells during lipolysis supplies the energy required for gluconeogenesis and provides ketone bodies, acetoacetate, and β-hydroxybutyrate, which can serve as alternative metabolic fuels for the central nervous system during periods of prolonged fasting. Studies have shown that an insulin infusion does not reduce hepatic glucose production if elevated levels of fatty acids are maintained by intravenous administration of a fat emulsion and heparin, suggesting that fatty acids may be the major mediator of gluconeogenesis.
The principal mechanisms regulating hepatic glucose output are the availability of gluconeogenic substrates and the regulation of transcription of key regulatory enzymes such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G-6-Pase). Glucagon and glucocorticoids through signaling intermediates induce the expression of PEPCK and G-6-Pase. Insulin on the other hand suppresses the expression of these enzymes.
Kidney: Although it is generally acknowledged that after fasting for 60 hours the kidney contributes up to 20% to 25% of endogenous glucose production, its role after an overnight fast remains controversial. One group has found that it produces as much as 25% of the postabsorptive glucose requirement, yet a second group using different methodology found a contribution of no more than 5% in the postabsorptive state. Despite these conflicting findings, it is clear that because the renal medulla removes almost as much glucose as the renal cortex produces, the net renal contribution is minimal in the short-term postabsorptive state as compared to the liver.
The kidney does not have glycogen stores and is dependent on gluconeogenesis as its only source of glucose production. Glutamine—rather than alanine—is the predominant amino acid substrate for renal gluconeogenesis. In addition to its contribution to glucose homeostasis after an overnight fast, the kidney also has been shown to be an important contributor to glucose counterregulation in the event of hypoglycemia. Although glucagon does not affect the kidney, the counterregulatory rise in catecholamines has been shown to stimulate gluconeogenesis in the renal cortex. As with the liver, insulin inhibits renal gluconeogenesis and glucose release. Hormonal changes that begin early in the postabsorptive state regulate the enzymatic steps necessary for hepatic glycogenolysis and hepatic and renal gluconeogenesis and ensure the delivery of the necessary substrate (Table 18–2). An appropriate fall in circulating insulin levels with a corresponding rise in glucagon is most important; elevations in the counterregulatory hormones, namely cortisol and growth hormone, contribute to it but are less critical. Thus, numerous endocrine and metabolic events interact to provide a continuous source of fuel for proper functioning of the central nervous system. Malfunction of any of these mechanisms can lead to symptomatic hypoglycemia.
| Decreased Insulin Secretion |
| Increases hepatic glycogenolysis |
| Increases lipolysis |
| Increases hepatic gluconeogenesis |
| Decreases muscle uptake of glucose |
| Increased Glucagon Secretion |
| Increases hepatic glycogenolysis |
| Facilitates hepatic gluconeogenesis |
| Increased Cortisol Secretion |
| Facilitates lipolysis |
| Increases protein catabolism |
| Augments hepatic gluconeogenesis |
Classification of Hypoglycemic Disorders
A clinical classification of the more common causes of symptomatic hypoglycemia in adults is presented in Table 18–3. This classification is useful in directing diagnostic considerations. Symptomatic fasting hypoglycemia is a serious and potentially life-threatening problem warranting thorough evaluation. Conditions that produce inappropriate fasting hyperinsulinism are the most common cause of fasting hypoglycemia in otherwise healthy adults. These include insulin-secreting pancreatic β cell tumors and iatrogenic or surreptitious administration of insulin or sulfonylureas. In patients with illnesses that produce symptomatic fasting hypoglycemia despite appropriately suppressed insulin levels, the clinical picture is generally dominated by the signs and symptoms of the primary disease, with hypoglycemia often only a late or associated manifestation. Hypoglycemia, sometimes severe, can also occur postprandially. In patients with gastric surgery, overrapid gastric emptying and accelerated glucose absorption may result in excess insulin secretion, rapid disposal of glucose, and hypoglycemia. An uncommon condition of islet hyperplasia or adult nesidioblastosis called noninsulinoma pancreatogenous hypoglycemia syndrome can result in hypoglycemia 4 to 6 hours after meals.
| Fasting |
| With hyperinsulinism |
| Insulin reaction |
| Sulfonylurea overdose |
| Surreptitious insulin or sulfonylurea self-administration |
| Autoimmune hypoglycemia (idiopathic insulin antibodies, insulin receptor autoantibodies) |
| Pentamidine-induced hypoglycemia |
| Pancreatic β cell tumors |
| Without hyperinsulinism |
| Severe hepatic dysfunction |
| Chronic renal insufficiency |
| Inanition |
| Hypocortisolism |
| Alcohol use |
| Nonpancreatic tumors |
| Nonfasting |
| Alimentary |
| Functional |
| Noninsulinoma pancreatogenous hypoglycemic syndrome (NIPHS) |
| Occult diabetes |
| Ethanol ingestion with sugar mixers |
Iatrogenic hypoglycemia is common in type 1 patients and also in insulin treated type 2 patients. Most type 1 patients aiming for HbA1c levels below 7% have on average one to two symptomatic hypoglycemic episodes per year. Severe hypoglycemia is defined as an episode requiring assistance, and in one study, incidence rates were about 12 per 100 patient years for both type 1 and insulin-treated type 2 patients. Sulfonylureas, repaglinide, and nateglinide can also cause hypoglycemia. Increased risk factors include age (70 years and older), renal failure, hepatic failure, and use of the long-acting sulfonylureas. A number of other drug-drug interactions (clarithromycin, salicylates, sulfonamides) can also potentiate the hypoglycemic effects of sulfonylureas. The annual incidence of sulfonylurea-induced hypoglycemia is approximately 0.2 per 1000 patient years.
As β cell failure progresses (early in type 1 and late in type 2 diabetes), patients lose their glucagon response to hypoglycemia. This combination of insulin deficiency and impaired glucagon response makes it harder for patients to achieve HbA1c levels below 7% without occasional hypoglycemia. These hypoglycemic episodes attenuate the sympathoadrenal response to hypoglycemia, with decreased epinephrine release from the adrenal and decreased sympathetic neural responses (hypoglycemic unawareness). The hypoglycemia unawareness in turn increases the risk for recurrent hypoglycemia. About 20% to 25% of type 1 patients have hypoglycemic unawareness. Other factors that increase the risk for hypoglycemia include poor self-management skills. Patients may take too much insulin for the carbohydrates or take the wrong insulin or do not appropriately time insulin administration with food ingestion. They also may not adjust the insulin for acute exercise or take extra carbohydrates for unexpected exercise or reduce insulin doses for improved insulin sensitivity with exercise training. Alcohol can decrease endogenous glucose production and can cause hypoglycemia, especially if it is consumed on an empty stomach. Diabetes complications—gastroparesis, autonomic neuropathy, and renal failure also increase the risk for hypoglycemia.
There are other consequences of hypoglycemia apart from the autonomic and neurogenic symptoms of acute hypoglycemia. In severe cases, hypoglycemia can cause convulsions and coma. Permanent neurological damage is rare. Although cross-sectional studies and case reports have reported intellectual decline with recurrent hypoglycemia, longitudinal studies have not shown significant cognitive dysfunction in adults. In the Diabetes Control and Complications Trial, there was no evidence for cognitive decline related to hypoglycemia in 18 years of follow-up. Young children however may be more vulnerable to the effects of hypoglycemia on the brain. Hypoglycemia via its autonomic stimulation and catecholamine release increases cardiac output. In patients with cardiac disease it can also precipitate cardiac arrhythmias, angina, myocardial infarction, and congestive heart failure.
Hypoglycemia can also exert a psychological toll. Acute hypoglycemia induces mood changes including fatigue, pessimism, anger, and behavior changes. Nocturnal hypoglycemia can lead to fatigue and decreased sense of well being the following day. Patients who have had severe hypoglycemia may develop a phobia about hypoglycemia and keep their sugars unreasonably high. Some patients develop an anxiety syndrome. Hypoglycemia can also impact personal relationships, occupation, driving, and leisure activities. Surveys show that type 1 patients have increased risk of driving mishaps (crashes, moving violations) when compared to nondiabetic spouses; and that these are related to hypoglycemia.
The goal of therapy is to restore levels of plasma glucose to normal as rapidly as possible. If the patient is conscious and able to swallow, glucose-containing foods such as candy, orange juice with added sugar, and cookies should be quickly ingested. Fructose, found in many nutrient low-calorie sweeteners for diabetics, should not be used. While it can be metabolized by neurons, fructose is not transported across the blood–brain barrier.
If the patient is unconscious, rapid restoration of plasma glucose must be accomplished by giving 20 to 50 mL of 50% dextrose intravenously over 1 to 3 minutes (the treatment of choice) or, when intravenous glucose is not available, 1 mg of glucagon intramuscularly or intravenously. Families or friends of insulin-treated diabetics should be instructed in the administration of glucagon intramuscularly for emergency treatment at home. Glucagon should not be given if the hypoglycemia is due to sulfonylurea use. Under these circumstances, glucagon can stimulate insulin secretion and worsen the hypoglycemia. Attempts to feed the patient or to apply glucose-containing jelly to the oral mucosa should be avoided because of the danger of aspiration. When consciousness is restored, oral feedings should be started immediately. In patients who have taken massive overdoses of sulfonylureas, the response to intravenous dextrose may be poor. For these patients, intravenous boluses of diazoxide (150-300 mg) may be tried but can result in hypotension. Intravenous octreotide (100 μg) has also been reported to be of benefit.
Patients on insulin or sulfonylureas should be instructed on how to recognize and treat hypoglycemia and what measures they can take to prevent such episodes. Type 1 patients and insulin-treated type 2 patients should monitor their blood glucose frequently. Hypoglycemia not infrequently occurs at night and patients should avoid taking large doses of short-acting insulin just before going to bed. Patients should from time to time also monitor blood glucose levels in the middle of the night. Hypoglycemia can also occur many hours after strenuous exercise, and patients should be advised to monitor at these times and cut back their insulin doses and/or eat more carbohydrate. Continuous glucose monitoring systems are increasingly used by type 1 patients to alert them to falling glucose levels and prevent hypoglycemia. Finally, it is important to individualize glycemic goals. Early in the course of both type 1 and type 2 diabetes when there is still some endogenous β cell function, it is easier to achieve HbA1c levels close to normal with low risk of hypoglycemia. As β cell failure progresses, however, aiming for normality may lead to unacceptably high rates of hypoglycemia. Patients who have had frequent hypoglycemia and have hypoglycemia unawareness should be encouraged to temporarily raise their glycemic targets—as little as 2 to 3 weeks of scrupulous avoidance of hypoglycemia reverses hypoglycemia unawareness and improves the attenuated epinephrine response. Diabetes complications, previous incidence of hypoglycemia, and life expectancy should all be considered in the establishment of glycemic goals.
Factitious hypoglycemia should be suspected in any patient with access to insulin or sulfonylurea drugs. It is most commonly seen in health professionals and diabetic patients or their relatives. The reasons for self-induced hypoglycemia vary, with many patients having severe psychiatric disturbances or a pathological need for attention. Inadvertent ingestion of sulfonylureas resulting in clinical hypoglycemia has also been reported, due either to patient error or to a prescription mishap on the part of a pharmacist. Patients with diabetes and factitious hypoglycemia have a presentation similar to brittle diabetes.
When insulin is used to induce hypoglycemia, an elevated serum insulin level often raises suspicion of an insulin-producing pancreatic β cell tumor. It may be difficult to prove that the insulin is of exogenous origin. The combination of hypoglycemia, high immunoreactive insulin levels, suppressed plasma C peptide, and suppressed proinsulin level is pathognomonic of exogenous insulin administration in nondiabetic patients. Patients with renal failure may have normal or even high plasma C-peptide levels, but plasma proinsulin levels are suppressed.
When sulfonylurea abuse is suspected, plasma or urine should be screened for its presence. Hypoglycemia with inappropriately elevated levels of serum insulin and C peptide along with detectable sulfonylureas in blood or urine is diagnostic of inadvertent or factitious sulfonylurea overdose. It is important to use assays which measure not only all the sulfonylureas but also repaglinide and nateglinide.
Treatment of factitious hypoglycemia involves psychiatric therapy and social counseling.
Numerous pharmacologic agents may potentiate the effects of insulin and predispose to hypoglycemia. Common offenders include fluoroquinolones such as gatifloxacin and levofloxacin, pentamidine, quinine, angiotensin-converting enzyme (ACE) inhibitors, ethanol, salicylates, and beta-adrenergic–blocking drugs. The fluoroquinolones, especially gatifloxacillin, have been associated with both hypoglycemia and hyperglycemia. Hypoglycemia is an early event; hyperglycemia occurs after several days into therapy. It is thought that the drugs act on ATP-sensitive potassium channels in the β cell. Intravenous pentamidine is cytotoxic to pancreatic β cells, resulting in acute insulin release and hypoglycemia. This occurs in about 10% to 20% of patients receiving the drug and may be followed later by persistent insulinopenia and hyperglycemia. Fasting patients taking noncardioselective beta blockade can have an exaggerated hypoglycemic response to starvation. Beta blockade inhibits fatty acid and gluconeogenic substrate release and reduces plasma glucagon levels resulting in hypoglycemia. Also the symptomatic response to hypoglycemia is altered—tachycardia is blocked while hazardous elevations of blood pressure may result during hypoglycemia in response to the unopposed alpha-adrenergic stimulation from circulating catecholamines and neurogenic sympathetic discharge. However, symptoms of sweating, hunger, and uneasiness are not masked by beta-blocking drugs and remain indicators of hypoglycemia in the aware patient.
Therapy with ACE inhibitors increases the risk of hypoglycemia in diabetic patients who are taking insulin or sulfonylureas, presumably because these drugs increase sensitivity to circulating insulin by increasing blood flow to muscle.
Ethanol-associated hypoglycemia has been proposed to occur as a consequence of hepatic alcohol dehydrogenase activity depleting NADH. The resultant change in the redox state (NADH-to-NAD+
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


