Abstract
Hypoglycemia is a disorder with manifold potential etiologies, from environmental to inherited, from presenting in isolation to presenting as a small part of a clinical syndrome, from insulin excess to defects in counterregulatory hormones or endogenous glucose production pathways, and from simple Mendelian disorders to those with more complex modes of inheritance, including imprinted conditions. Classic neuroglycopenic and adrenergic symptoms may be absent in infants, where hypoglycemia often presents with nonspecific symptoms. Diagnosis requires evaluating the response to hypoglycemia by means of obtaining a critical sample at the time of hypoglycemia, whether spontaneous or elicited by a diagnostic fast; however, a diagnostic fast should never be undertaken without first excluding the possibility of a fatty acid oxidation defect via an acylcarnitine profile. Molecular genetic testing can confirm the underlying genetic diagnosis. Hypoglycemia treatment is specific to the cause of the hypoglycemia, which is why proper diagnosis is crucial.
Keywords
hypoglycemia, insulin, counter-regulatory hormones, endogenous glucose production
Introduction
Overview of Glucose Homeostasis
Glucose is the primary metabolic fuel – that is, the main substrate for energy metabolism. Thus, maintaining its levels in an optimal range is crucial for health and survival, and is regulated by a complex interplay of metabolic processes controlling glucose utilization and glucose production. The key metabolic pathways involved in the glucose utilization in the fed state are glycolysis (the breakdown of glucose to generate ATP and energy) and the Krebs cycle, also known as the citric acid cycle. The key anabolic pathways ensuring that excess glucose is stored and can later be utilized for energy are glycogen synthesis, protein synthesis, and lipogenesis; these pathways are stimulated by the actions of insulin. In addition, insulin causes the translocation of the GLUT4 glucose transporters from the cytosol to the cell membrane of myocytes and adipocytes to increase glucose uptake.
Endogenous glucose supply in the fasting state depends on the recognition of dropping glucose levels and consequent suppression of insulin and triggering of the hormonal counterregulatory response, stimulating the following key endogenous glucose production pathways:
- •
Glycogenolysis: Breakdown of hepatic glycogen. This suffices to maintain euglycemia from several hours in a child to up to 12 h in an adult).
- •
Gluconeogenesis: The generation of glucose from noncarbohydrate precursors – primarily lactate, pyruvate, amino acids (principally alanine and glutamine), and glycerol (from fat). Once the glycogen supply is exhausted, gluconeogenesis becomes the predominant glucose production pathway until its supplies are exhausted.
- •
Lipolysis: The generation of free fatty acids and glycerol from triglycerides (triacylglycerol).
- •
Fatty acid oxidation and ketogenesis: The generation of acetoacetate and beta-hydroxybutyrate through β-oxidation from free fatty acids.
These processes, discussed further, normally help to assure that sufficient glucose is available to all body tissues to fulfill their metabolic needs; a defect in one or more of these can lead to hypoglycemia. The endogenous glucose production pathways are suppressed by insulin and stimulated by the counterregulatory hormones, namely, glucagon, epinephrine, cortisol, and growth hormone (GH). Equilibrium is maintained by the central nervous system (CNS), which integrates systemic signaling and helps upregulate or suppress insulin and the counterregulatory responses, as well as coordinating the behavioral, neuroendocrine, and autonomic responses to hypoglycemia. A schema of the counterregulatory hormone response to hypoglycemia is illustrated in Fig. 3.1 .
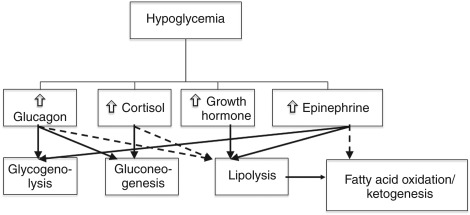
Background and Incidence/Prevalence
The definition of hypoglycemia is controversial, especially in neonates and infants, where operational thresholds for abnormally low glucoses range from 18 mg/dL to 70 mg/dL (1–4 mmol/L). An ideal operational definition for neonatal hypoglycemia would be defined as the blood glucose concentration below which value significant morbidity (especially neurologic sequelae) would be seen and therefore at which intervention should be undertaken. However, the plasma glucose level and duration of hypoglycemia associated with adverse neurodevelopmental sequelae has not been conclusively established. It should be also borne in mind that hypoglycemia on the day of birth or during the first few days of life may not necessarily have a pathological etiology. The transition between intrauterine and extrauterine life involves an inherent breaking off of the previously continuous supply of maternal glucose, and the consequent need for the neonate to adapt to fasting. This transition is marked by a number of other changes, including a 3–10-fold surge in catecholamine secretion at the time of delivery; a 3–5-fold increase in glucagon concentration, with a concomitant elevation in GH and cortisol; and, under ordinary circumstances, a drop in insulin secretion. All of these endocrine changes facilitate activation of pathways of fasting adaptation – namely, glycogenolysis, gluconeogenesis, lipolysis, fatty acid oxidation, and ketogenesis. It is noteworthy that these fasting mechanisms are not mature at the time of birth; therefore, a transient drop in blood glucose levels one to two h after birth is common, even as low as 30 mg/dL, and does not necessarily lead to adverse sequelae. It is the infants with persistent hypoglycemia who are of concern and who must be evaluated for potential genetic etiologies.
Because different definitions of hypoglycemia are in use and there is no consensus definition, true incidence and prevalence of neonatal hypoglycemia is difficult to determine. Prevalence also depends upon the population in question – in at-risk groups, such as premature infants, small for gestational age infants, infants of diabetic mothers, or exclusively breastfed infants, prevalence of hypoglycemia in the first three days of life may be as high as 51% without necessarily indicating a genetic etiology. Prevalence of asymptomatic hypoglycemia (defined as blood glucose <45 mg/dL) in normal neonates in the first days of life may be as high as 5–15%. In a more recent case–control study of hypoglycemia incidence in full-term neonates weighing more than 2500 g that were products of nondiabetic, singleton pregnancies, using a cutoff for hypoglycemia of below 50 mg/dL, hypoglycemia prevalence in the first 24 h was 2.4%. In this study, hypoglycemia was found to be more common in infants whose mothers had puerperal fever, public versus private insurance, and/or who were earlier in gestation. It should be noted that serial glucose levels were not measured in this study, and that the true prevalence of hypoglycemia could have been underestimated. Although the prevalence estimates vary, by all measures, neonatal hypoglycemia is relatively common and, if persistent, can lead to devastating sequelae, including seizures, visual disturbances, motor and speech delay, cerebral palsy, and white matter injury. Thus, it is as incumbent upon care providers of infants to recognize this clinical entity and perform the appropriate diagnostic evaluation as it is upon care providers of older children and adults. Hypoglycemia has many different potential etiologies, ranging from inborn errors of metabolism to deficiencies in counterregulatory hormones, insulin excess, and many others. In addition to inborn errors, there are also a large variety of environmental exposures and other potential noninherent causes of hypoglycemia to consider in the differential diagnosis (e.g., exogenous insulin or insulin-sensitizing agents, salicylate poisoning, and others); while this chapter focuses only on inborn genetic and epigenetic entities causing hypoglycemia, the existence of a wider differential diagnosis should not be forgotten. Finally, it should be borne in mind that although many hypoglycemic disorders of genetic etiology manifest in the neonatal period, not all do so; some only manifest with more prolonged fasting in later infancy and the toddler years, and some mild disorders may not manifest until adulthood.
Clinical Presentation
Clinical presentation of hypoglycemia varies by age. In general, symptoms of hypoglycemia may be divided into two categories, adrenergic and neuroglycopenic. More about the different symptoms can be found in the following list, along with a separate discussion of presentation in infants:
- 1.
Adrenergic symptoms : Sweating, pupillary dilatation, tachycardia, shakiness or tremors, anxiety, nervousness, hunger, nausea/emesis.
- 2.
Neuroglycopenic symptoms : Dizziness, confusion, lethargy, headache, visual disturbances, difficulty concentrating, speech difficulty, somnolence/prolonged sleep, hypothermia, seizures or twitches, personality changes, temper outbursts, bizarre or manic behavior, depression or psychosis, and at the extreme, loss of consciousness and coma, with risk of permanent neurological damage or death.
- 3.
Presentation in neonates : This requires a higher index of suspicion, because symptoms in infants may not be specific to hypoglycemia. In neonates and infants, symptoms and signs may include poor feeding or feed refusal, cyanosis or apneas, somnolence or lethargy, seizures or myoclonic jerks, emesis, sweating, low temperatures, and hypotonia.
Beyond these general symptoms, the clinical presentation of hypoglycemia varies by the underlying disorder, and will thus be discussed separately under each disorder in the subsequent sections.
Diagnostic Evaluation
The initial part of hypoglycemia evaluation lies in obtaining a history of the circumstances – fasting versus postprandial hypoglycemia, duration of fast required that elicits the hypoglycemia, associated symptoms, clinical circumstances (whether associated with intercurrent illness or other catabolic stress or not), and other associated clinical features. Nongenetic causes of hypoglycemia (e.g., salicylate or alcohol intoxication, exogenous insulation, or sulfonylurea exposure) should also be considered. Physical examination should be performed to examine for potential clinical symptoms that would suggest an etiology, such as hepatomegaly, which is common in glycogen storage diseases (GSDs), or characteristic dysmorphisms seen in certain syndromes associated with hypoglycemia such as Beckwith–Wiedemann syndrome. An acylcarnitine profile should be obtained to screen for disorders of fatty acid oxidation; individuals with such disorders should not undergo a diagnostic fast, or else a metabolic crisis may be triggered. Total and free carnitine levels should also be sent, as individuals with carnitine deficiency will have hypoglycemia, and these levels are not affected by fasting.
The most critical component of the hypoglycemia evaluation is the obtaining of a critical sample of blood and urine at the time of hypoglycemia (serum glucose <50); the hypoglycemia may be spontaneous, or may need to be elicited through a diagnostic fast of age-appropriate length. The diagnostic sample should include the following labs: serum glucose, insulin, C-peptide, CO 2 , beta-hydroxybutyrate, free fatty acids, lactate, ammonia, GH, cortisol, serum amino acids, and urine organic acids. Finally, after a critical sample has been obtained, a glucagon stimulation test should be performed as further evaluation for insulin-mediated hypoglycemia: 1 mg glucagon is administered intravenously, and glucose levels are checked every 10 min for 40 min. A positive response to the glucagon stimulation test is a rise in glucose of greater than 30 mg/dL in 20 min or 40 mg/dL in 40 min.
Genetic pathophysiology
Hyperinsulinism
Hyperinsulinism (HI), also known as hyperinsulinemic hypoglycemia, is a disorder characterized by inappropriate insulin secretion by pancreatic β-cells, resulting in mild to severe hypoglycemia. This is then divided into two categories. The first is nongenetic perinatal stress-induced HI, a poorly understood disorder in which infants exposed to prenatal or perinatal stress such as preeclampsia, hypertension, perinatal asphyxia, prematurity, or intrauterine growth retardation manifest transient hyperinsulinemic hypoglycaemia, which self-resolves (median time to resolution is 6 months ). The second is congenital HI, genetically and clinically heterogeneous, which results from various mutations along the insulin secretory pathway – primarily involving genes that encode the ATP-sensitive potassium (K ATP ) channel involved in insulin secretion as well as enzymes involved insulin secretion or, less commonly, gain of function mutations, which give rise to aberrant functioning proteins, which can impact insulin secretion. Fig. 3.2 illustrates normal glucose stimulated insulin secretion and the various mutations that can lead to HI, as well as the sites action for potentially therapeutic agents diazoxide and octreotide (a somatostatin) and, conversely, of sulfonylurea medications.
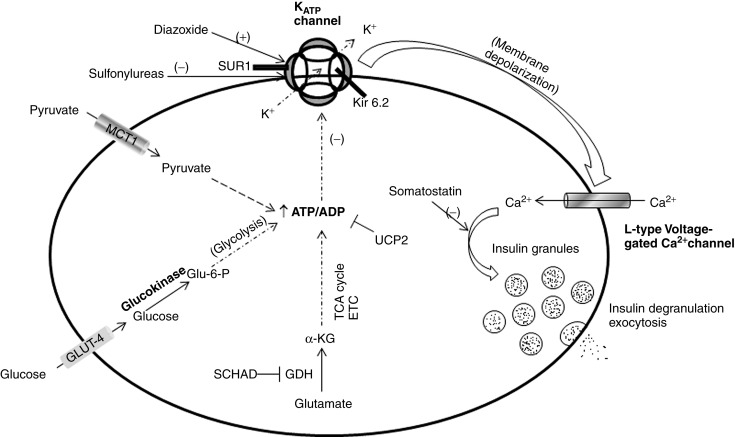
The prevalence of congenital HI is estimated to be between 1/30,000 and 1/50,000, although the prevalence is higher in some populations, from ∼1/11,000 in Ashkenazi Jews to as high as 1/2500 in Saudi Arabia. The most common mutations are in the ABCC8 and KCNJ11 genes, which respectively encode the SUR1 and Kir6.2 subunits of the β-cell plasma membrane K ATP channel. In a recent study of 417 children with HI, 91% of diazoxide-unresponsive patients had identifiable mutations (89% in either ABCC8 or KCNJ11 , and 2% in GCK , which encodes glucokinase, the enzyme that controls the rate-limiting step in glycolysis within the β-cell). Of patients with diazoxide-responsive HI, 47% had identifiable mutations (42% in GLUD1 , which encodes glutamate dehydrogenase,41% were dominant K ATP channel mutations, and 16% were rare gene mutations). Genetic defects leading to HI are outlined in Table 3.1 and discussed further in the subsequent section.
| HI type | Hyperinsulinemic hypoglycemia, familial # | Gene/s | Affected protein/enzyme | Chromosomal location | Inheritance | Clinical features | Treatment |
|---|---|---|---|---|---|---|---|
| |||||||
| Hyperinsulinism relating to mutations in the KATP channel (K ATP -HI) | HHF-1 (ABCC8) HHF2 (KCNJ11) | ABCC8 KCNJ11 | SUR1 ( ABCC8 ) or Kir6.2 ( KCNJ11 ) | 11p15.1 11p15.1 | Diffuse: autosomal recessive Focal: sporadic. “2-hits”: inheritance of paternal mutation + loss of maternal heterozygosity |
|
|
| Dominant K ATP -HI | ABCC8 KCNJ11 | SUR1 or Kir6.2 | 11p15.1 11p15.1 | Autosomal dominant | Milder hypoglycemia than recessive K ATP -HI
| Diazoxide-responsive | |
| Hyperinsulinism- hyperammonemia (HI-HA, or GDH-HI) | HHF-6 | GLUD1 | GDH | 10q23.3 |
|
| Diazoxide-responsive |
| Glucokinase-hyperinsulinism (GCK-HI) | HHF-3 | GCK | Glucokinase (hexokinase 4) | 7p15.3–7p15.1 | Autosomal dominant; may also be de novo |
| Diazoxide-responsive in most cases; may require subtotal pancreatectomy in severe cases |
| SCHAD-HI | HHF-4 | HADH | Hydroxyacyl dehydrogenase | 4q25 | Autosomal recessive | Rare. Hypoglycemia ranges from mild to severe; abnormal acylcarnitine profile | Diazoxide-responsive |
| MCT1 (exercise-induced HI, or EIHI) | HHF-7 | SLC16A1 | Proton-linked monocarboxylate transporter, member 1 (MCT-1) | 1p13.2 | Autosomal dominant | Exercise-induced hyperinsulinemic hypoglycemia | Carbohydrate intake during exercise; limit anaerobic exercise |
| HNF4A-HI | HNF4A | Hepatocyte nuclear factor 4α | 20q13.12 | Autosomal dominant |
| Diazoxide-responsive | |
| |||||||
| INSR-HI | HHF-5 | INSR | Insulin receptor | 19p13.2 | Autosomal dominant | Postprandial hypoglycemia, fasting hyperinsulinemia | Long-active somatostatin analog |
| HNF1A-HI | HNF1A | Hepatocyte nuclear factor 1α | 12q24.2 | Autosomal dominant | Rare | Diazoxide-responsive, partially | |
| UCP2-HI | UCP2 | Uncoupling protein 2 | 11q13.4 | Autosomal dominant | Rare. Tends to self-resolve within a few years | Diazoxide-responsive | |
| HK1 | Hexokinase 1 | 10q22.1 | Autosomal dominant (in some cases, somatic mosaicism) | Rare | Diazoxide-responsive. Somatic mutations with focal lesions can be cured by partial pancreatectomy. | ||
| KCNQ1 | Kv7.1 | 11p15.5 | Autosomal dominant | One case series. Prolonged QT interval; postprandial hyperinsulinemic hypoglycemia; hypokalemia after oral glucose load | Unknown if diazoxide-responsive | ||
| AKT2 | AKT2 – part of the insulin receptor signal transduction cascade | 19q13.2 | Autosomal dominant | Two case reports
| Diazoxide unresponsive; require regular carbohydrate feeds to maintain euglycemia | ||
Hyperinsulinism Resulting from Mutations in ABCC8 or KCNJ11 (Hyperinsulinemic Hypoglycemia, Familial Types 1 and 2) – K ATP -HI
ABCC8 and KCNJ11 encode SUR1 and Kir6.2, respectively, which together form the subunits of the heterooctameric K ATP channel in the β-cell plasma membrane. This channel is comprised of four inner Kir6.2 subunits, which form the channel’s ion pore, and four regulatory SUR1 subunits, which surround the Kir6.2 core. These subunits are assembled in the endoplasmic reticulum. Normal insulin secretion results from a fuel stimulated increase in the ATP/ADP ratio within the β-cell leading to closure of the K ATP channel with resultant depolarization of the β-cell membrane and opening of a voltage-gated calcium channel with subsequent Ca 2+ influx. This influx leads to insulin degranulation and exocytosis. Inactivating mutations of either the K ATP or SUR1 moieties of the K ATP channel result in reduced or absent K ATP channel activity, leading to permanent depolarization, spontaneous and independent opening of the voltage-gated Ca 2+ channels, and consequent constitutive insulin secretion irrespective of serum glucose.
Over 300 mutations in ABCC8 and over 30 mutations in KCNJ11 have been described in association with HI. Mutations may lead to an inability of the altered channel to exit the endoplasmic reticulum and be trafficked to the cell membrane, a disruption of the nucleotide-binding domain of SUR1 (i.e., lack of response to increased Mg-ADP concentrations ), an alteration of K ATP channel density, or a complete abolishment of channel activity.
Recessively inherited K ATP -HI is both the most common and most severe form of HI. Clinically, children present with severe hyperinsulinemic hypoglycemia, often, though not always, are in the neonatal period. The initial medical management for a child diagnosed with HI is with diazoxide, a medication that acts on the K ATP channel to prevent closure of the channel. Diazoxide unresponsive HI suggests a K ATP channel defect and a potential need for surgery. Genetic testing of the child and parents is recommended to further delineate the type of HI. K ATP -HI is divided into two histopathological morphologies: diffuse disease and focal disease. Diffuse K ATP -HI is, in most cases, due to autosomal recessive mutations in ABCC8 or, less commonly, KCNJ11 .
Children with a homozygous or compound heterozygous recessive mutation will have diffuse disease, whereas children with a paternally inherited heterozygous recessive mutation most likely have focal disease. Focal HI occurs as a result of inheriting a paternally derived mutation in ABCC8 or KCNJ11 and experiencing a somatic loss of the maternal 11p15 allele, resulting in paternal uniparental isodisomy. The maternal loss of heterozygosity entails loss of tumor suppressor genes, including CDKN1c [p57(kip2)] and H19; there is also upregulation of paternal IGF2. The combination of loss of tumor suppressor genes and upregulation of IGF2 leads to adenomatous expansion of the affected region. Thus, somatic regions within the pancreas in which a paternal mutation has been inherited and maternal loss of heterozygosity has occurred become foci of HI (focal adenomatosis). While focal and diffuse HI resemble each other clinically much of the time, a recent article reviewed 223 children with diffuse or focal HI and reported several significant clinical differences between the two (all values in parentheses are means and all differences reached statistical significance). Compared to children with focal disease, children with diffuse disease were born at an earlier age (38 weeks vs. 39 weeks) and were born with larger birth weights (3,963 vs. 3,717 g). Additionally, children with diffuse HI had higher insulin levels at the time of diagnosis (31.8 μU/mL vs. 12 μU/mL) and had higher maximal GIR requirements (19.2 mg/kg/min vs. 16.1 mg/kg/min). Children with focal HI presented at a later age compared to those with diffuse disease (0.3 months vs. 0 months) and were more likely to present with a seizure (50% vs. 25%). Distinguishing focal from diffuse disease is of utmost importance as focal disease can be cured by surgical resection of the lesion, whereas diffuse disease requires a near-total pancreatectomy. In addition to genetic testing, an F-DOPA PET scan can help in localization of a focal lesion. In the review of 223 patients, those with diffuse disease had a median percent pancreatectomy of 98%, compared to 27% in those with focal disease. Of the children that had surgery for diffuse disease, 41% had persistent hypoglycemia requiring treatment with enteral dextrose and/or octreotide. Comparatively, 94% of the children that had surgery for focal disease required no further management.
Autosomal dominant mutations in KCNJ11 and ABCC8 have been reported. They are characterized by normal subunit trafficking but impaired K ATP channel activity, which overall can result in a milder hypoglycemia compared to the recessive mutations earlier, and many are responsive to therapy with diazoxide, unlike the recessively inherited mutations.
Hyperinsulinism Resulting from Mutations in GLUD-1 (Hyperinsulinemic Hypoglycemia, Familial 6) – GDH-HI
GLUD1 encodes glutamate dehydrogenase (GDH), a mitochondrial matrix enzyme that catalyzes the oxidative deamination of glutamate to alpha-ketoglutarate and ammonia with the assistance of either NAD + or NADP + as a cofactor. GDH is an important regulator of amino acid and ammonia metabolism; it is expressed at high levels in the brain, hepatocytes, pancreatic β-cells, and the kidneys, but not in muscle. GDH enzyme activity is allosterically inhibited by ATP and GTP and allosterically activated by ADP, GDP, and leucine.
GDH-HI results from gain-of-function mutations in GLUD1. The insulin secretion may be triggered by glucose or by amino acids, and is potentiated by leucine. Eighty percent of GDH-HI cases are due to de novo mutations in GLUD1; the remaining 20% are inherited in an autosomal dominant pattern. Over 20 disease-causing mutations have been identified. Most heterozygous missense amino acid substitutions in GLUD1 affect the GTP-binding site, decreasing the sensitivity of GDH to inhibition by GTP to varying degrees, increased sensitivity to leucine stimulation, and leading to excess insulin secretion; the degree of sensitivity to GTP inhibition is inversely related to severity of hypoglycemia phenotype. Other mutations affect the antenna region of GDH, which communicates with adjacent enzyme subunits. Increased GDH activity results in increased glutamate oxidation to alpha-ketoglutarate, which enters the Krebs cycle and leads to increased ATP levels. The increased ATP/ADP ratio leads to K ATP channel closing and insulin secretion as discussed earlier. The mutations result in increased enzyme activity in pancreatic β-cells, hepatocytes, in the kidneys, and in the brain. These children also manifest asymptomatic persistent elevations in serum ammonia levels due to increased hepatic GDH activity (and with some contribution from the kidney ); thus, this syndrome is referred to as hyperinsulinism-hyperammonemia syndrome, or HI-HA.
Clinically, children present recurring episodes of both fasting and postprandial hypoglycemia, especially after consuming protein-rich meals. This hypoglycemia is typically milder than that seen in K ATP -HI and is thus often not diagnosed at birth. It is responsive to diazoxide. Although ammonia levels are usually (though not always) quite elevated, typically two to five times above the usual normal limits, affected patients typically do not manifest hyperammonemia symptoms. That said, it should be noted that generalized seizures, especially absence seizures, may be seen in patients with GDH-HI even without concurrent hypoglycemia; this may be due to hypoglycemic brain damage, chronically elevated ammonia levels, or CNS hyperexcitability due to GDH mutations. The exact etiology of the seizures, however, is unclear. Additionally, children with HI-HA have a higher incidence of developmental delays and behavior problems.
Hyperinsulinism Resulting from Mutations in GCK (Hyperinsulinemic Hypoglycemia, Familial 3) – GK-HI
GCK encodes glucokinase (GK), an isoform of hexokinase expressed in pancreatic β-cells, hepatocytes, glucose-sensing hypothalamic neurons (predominantly in the ventromedial nucleus and arcuate nucleus), and some enteroendocrine cells. GK acts as a glucose sensor and catalyzes the initial and rate-limiting step in glycolysis, the conversion of D -glucose to D -glucose-6-phosphate at physiologic glucose concentrations using MgATP as a second substrate. GK exists in three different conformations, which control catalytic function, generating a sigmoidal response to plasma glucose concentrations; the transitions between these conformations are controlled by glucose concentrations and by allosteric interactions with modulators of GK activity such as biotin, cyclic AMP, and insulin, as well as glucokinase regulatory protein (GKRP). GK acts as the β-cell’s glucose sensor because it has a relatively low affinity for its substrate, glucose; thus, it determines the glucose threshold for insulin release. The typical set-point for glucose-induced GK expression in β-cells is ∼5 mmol/L, or 72 mg/dL.
Autosomal dominant activating mutations in GCK are relatively rare causes of congenital HI; as of this writing, only 12 activating mutations have been identified. That said, one recent multicenter study found that GCK mutations were responsible for up to 1.2% of cases of congenital HI and nearly 7% of non-K ATP -channel cases of HI. These activating missense mutations typically occur in or near the allosteric binding sites for either GK activators or the competitive inhibitor, GKRP; the net result of activating mutations is to increase the affinity of GK for its substrate glucose, effectively lowering the threshold for insulin release, sometimes to as low as 1 mmol/L or 9 mg/dL. Of interest, mutations that conversely decrease GK’s binding affinity for glucose and increase the threshold for insulin secretion are responsible for a form of monogenic diabetes, MODY2; it is noteworthy that inactivating mutations are far more numerous than activating GCK mutations.
Clinically, the phenotype of GK-HI ranges from severe neonatal HI to mild fasting hypoglycemia, which may not be recognized until later in childhood or even into adulthood. Many patients will respond to diazoxide, but the degree of responsiveness can be quite variable. Patients may demonstrate an initially good response, but over time become less responsive. Those that do not respond to diazoxide or lose responsiveness over time may require a partial or near-total pancreatectomy in order to treat the HI.
Hyperinsulinism Resulting from Mutations in HADH (Hyperinsulinemic Hypoglycemia, Familial 4) – SCHAD-HI
HADH encodes medium/short-chain-3-hydroxyacyl-CoA dehydrogenase (SCHAD), a mitochondrial matrix enzyme that catalyzes the third and penultimate step in β-oxidation of fatty acids into acetyl-CoA, the conversion of 3-hydroxyacyl-CoA to 3-ketoacyl-CoA using NAD + as a cofactor. There are different isoforms of the various enzymes that catalyze the β-oxidation of medium- and short-chain fatty acids. In addition to its actions in the β-oxidation pathway, SCHAD also acts as an allosteric inhibitor of GDH, resulting in decreased affinity of GDH for its substrate, exerting an overall inhibitory effect on GDH in pancreatic islets resulting in excessive insulin secretion.
HADH loss-of-function mutations are a very rare cause of HI, although they are more common in children born to consanguineous parents and in children of Irish or Turkish populations, wherein there is a founder mutation. These mutations have an autosomal recessive inheritance mode. Several mutations have now been reported. The mechanism by which SCHAD mutations cause hyperinsulinemic hypoglycemia (as opposed to noninsulin mediated hypoketonemic hypoglycemia) entails loss of its inhibitory effect on GDH, leading to increased GDH affinity for leucine, alanine, and glutamate. Mechanisms by which the mutations cause SCHAD loss of function include causing deletions or alternate splice variants within the gene, mutations affecting protein folding, or deep intronic mutations introducing a frameshift in the gene.
Clinically, children present with hyperinsulinemic hypoglycemia ranging from mild to severe in degree, accompanied by elevated levels of plasma 3-hydroxybutyrylcarnitine in both the fasting and fed states and elevated urine 3-hydroxyglutaric acid. Unlike most other fatty acid oxidation disorders, children with SCHAD-HI typically do not have myopathies of skeletal and/or cardiac muscle and also typically do not have hepatotoxicity. SCHAD-HI is typically responsive to diazoxide.
Hyperinsulinism Resulting from Mutations in SLC16A1 (Hyperinsulinemic Hypoglycemia, Familial 7) – Exercise-Induced-HI
Proton-linked monocarboxylate transporter, member 1 (MCT-1) is an enzyme that catalyzes the movement of several monocarboxylates (e.g., lactate, pyruvate, valine, leucine-derived branched-chain oxo-acids, and ketone bodies) across the plasma membrane. It is widely expressed across multiple tissues, but its expression in pancreatic α- and β-cell membranes is typically low to absent. In addition, expression of lactate dehydrogenase-A, which is responsible for the conversion of pyruvate to lactate as the final step of anaerobic metabolism, is relatively low in β-cells; this helps channel pyruvate preferentially toward mitochondrial oxidative metabolism rather than toward lactate, and keeps glucose rather than pyruvate or lactate as the preferential trigger for insulin secretion. As a result of the low β-cell MCT-1 expression and LDH-A activity, MCT-1 activity does not ordinarily contribute to β-cell insulin secretion – in other words, changes in extracellular concentrations of lactate or pyruvate (e.g., during exercise or during a catabolic state) do not trigger insulin secretion.
In exercise-induced hyperinsulinism (EIHI, or MCT-HI), a gain-of-function mutation in the regulatory regions of SLC16A1 in the binding sites of several transcription factors allows for unusually high levels of expression of MCT-1 in β-cell membranes, enabling β-cell pyruvate uptake and pyruvate-stimulated insulin release in the presence of elevated lactate or pyruvate levels, such as in the setting of intense exercise or anaerobic exercise, leading to triggering of the usual K ATP -mediated insulin secretory pathway and thus to ensuing hypoglycemia. Three different autosomal dominant mutations have been identified – two in Finnish families and one in a German family.
Clinically, patients present with hyperinsulinemic hypoglycemia of varying degrees in the setting of exercise; response to diazoxide is partial. Treatment involves limiting anaerobic exercise and frequent intake of carbohydrate-containing snacks during aerobic exercise.
Hyperinsulinism Resulting from Mutations in INSR (Hyperinsulinemic Hypoglycemia, Familial 5) – Insulin Receptor Activating Mutations
INSR encodes the insulin tyrosine kinase receptor, which is a heterotetrameric protein composed of two extracellular α subunits to which insulin binds and two transmembrane kinase β subunits. The binding of insulin to the α subunits stimulates the kinase, which triggers autophosphorylation of the β subunits on tyrosine residues, which in turn leads to the phosphorylation of a number of intracellular substrates, including the insulin receptor substrates. These phosphorylated substrates in turn activate two signaling cascades: the phosphotydilinositol-3-kinase-AKT/protein kinase B pathway, which mediates the majority of insulin’s metabolic actions, and the Ras-mitogen-activated protein (MAP) kinase pathway.
In 2004, an activating, autosomal dominant mutation in INSR was reported in three generations of a Danish family. This was a missense mutation in the tyrosine kinase domain of the insulin receptor gene. This is the only reported mutation in INSR linked to hypoglycemia – all other known pathogenic mutations cause variable degrees of insulin resistance and a diabetic phenotype.
Affected children and adults present with fasting hyperinsulinemia, elevated insulin: C-peptide ratios, and moderate to severe postprandial hypoglycemia coexisting in some cases with impaired glucose tolerance (first a significant rise in blood glucose levels postglucose challenge, then a lower-than-normal nadir). All affected patients manifested insulin resistance as measured by hyperinsulinemic-euglycemic clamp. Thus, the mutation caused a divergent phenotype of insulin resistance and insulin-mediated hypoglycemia. Patients responded to treatment with a long-acting formulation of octreotide, a somatostatin analog that acts downstream of the voltage-gated calcium channel in the β-cell to inhibit insulin secretion.
Hyperinsulinism Resulting from Mutations in HNF1A (Hepatocyte Nuclear Factor-1α)
HNF1A encodes the protein HNF-1α, a transcription factor required for the expression of several liver-specific genes. More specifically, the homodimer protein binds to a promoter sequence that is required for hepatocyte-specific transcription of hepatic-specific genes, including fibrinogen, alpha-1-antitrypsin and others. HNF-1α’s dimerization affects gene transcription.
HNF1A mutations are typically associated with a form of autosomal dominant monogenic diabetes (MODY3); however, in 2011, one case was described where an adult with MODY3 was found to have had fetal macrosomia and childhood hypoglycaemia, which self-resolved prior to developing diabetes mellitus at the age of 19. This mutation was heterozygous (implying dominant inheritance) and affected the HNF1A gene’s DNA-binding domain. Two other mutations have since been reported, one a heterozygous nonsense mutation affecting HNF1A ’s dimerization domain and another a heretozygous missense mutation. The mechanism by which loss of function mutations in HNF1A lead to hypoglycemia alone or the dual phenotype of hypoglycemia in infancy/childhood and monogenic diabetes in adulthood has not been well elucidated.
Clinically, the reported hypoglycemia phenotypes range from isolated incidents in childhood to full-blown neonatal HI. Variable degrees of responsiveness to diazoxide have been reported.
Hyperinsulinism Resulting from Mutations in HNF4A (Hepatocyte Nuclear Factor 4-Alpha)
HNF4A encodes the protein HNF4-α, a nuclear transcription factor that binds DNA (to HNF4-α response elements and with the involvement of coactivator peptides) as a homodimer. HNF4-α is the most abundant hepatic DNA-binding protein, influencing the expression of approximately 40% of actively transcribed hepatic genes, including HNF1A and other genes involved in gluconeogenesis and hepatic liver metabolism. HNF4-α is also expressed in the pancreas, and controls the expression of approximately 11% of pancreatic islet genes.
Most pathogenic mutations in HNF4A cause monogenic diabetes (MODY1). Heterozygous loss-of-function mutations in HNF4A have also been linked with hyperinsulinemic hypoglycemia. Most of these mutations affect the DNA-binding domain; some affect the ligand-binding domain. In one recent series, HNF4A mutations were found to be the third most common mutations to cause diazoxide-responsive HI – mutations were seen in 11/220 cases, with each case being caused by a different mutation. Inheritance is generally autosomal dominant, though de novo mutations were seen in 4/11 of the cases above. MODY1 and the hyperinsulinemic hypoglycemia may not be entirely unrelated – in one report, ∼14% of patients with MODY1 had a dual phenotype, having experienced transient hypoglycemia in infancy, in addition to fetal macrosomia (which was also reported in a larger subset of patients with HNF4A mutations but without hypoglycemia ).
Clinically, affected children often manifest fetal macrosomia. The infantile hyperinsulinemic hypoglycemia has a range of severity, from mild and transient hypoglycemia requiring brief intravenous glucose support to more severe, persistent hypoglycemia requiring pharmacotherapy for several years. Cases of HNF4A-HI are typically responsive to diazoxide. One reported case also developed hypophosphatemic rickets, renal Fanconi syndrome, and hepatic glycogenosis. HNF4A mutations may have a dual phenotypic manifestation – patients experiencing infantile hypoglycemia are at risk of developing MODY1, a form of monogenic diabetes, as described earlier.
Hyperinsulinism Resulting from Mutations in UCP2 (Uncoupling Protein 2)
UCP2 encodes a mitochondrial uncoupling protein that facilitates anion transfer from the inner to the outer mitochondrial membrane in exchange for protons. It is part of a larger family of uncoupling proteins that separate (“uncouple”) oxidative phosphorylation from ATP synthesis; the resulting energy is dissipated as heat. UCP2 is widely expressed in many tissues, including pancreatic islets. UCP2 acts as a negative regulator of insulin secretion in pancreatic β-cells by decreasing ATP content and thus inhibiting glucose-stimulated insulin secretion. Two heterozygous, autosomal dominant loss-of-function UCP2 mutations have been reported to cause congenital HI. Both cases were diazoxide responsive.
Other Rare Forms of Hypoglycemia Due to Hyperinsulinemia or Related Causes
- 1.
Hexokinase 1 ( HK1 ): Hexokinase 1 is another member of the mitochondrial hexokinase family, like glucokinase; it catalyzes the first step in glycolysis, namely, the conversion of glucose into glucose-6-phosphate. Several cases of fasting and postprandial HI due to autosomal dominant HK1 mutations have been reported in a single family. All cases presented with moderate to severe hypoglycemia in infancy; 40% of those presented with a seizure. All responded well to diazoxide treatment. In another report, a form of HI involved islet cell mosaicism, with some localized hyperfunctioning islets with low insulin storage along with high proinsulin production, and a larger number of atrophied islets; the hyperfunctioning was found to be due to either elevated expression of low-Km HK1 (which is normally expressed at minimal levels in β-cells) substituting for GCK in β-cell glycolysis, or to an activating mutation in GCK . These cases of HI responded to diazoxide and/or were cured by focal pancreatectomy.
- 2.
KCNQ1: KCNQ1 encodes a voltage-gated potassium channel with six transmembrane regions. It is located on a region of chromosome 11 along with a cluster of other genes involved in Beckwith–Wiedemann syndrome. KCNQ1 is primarily found in the plasma cell membrane of both cardiac myocytes, where activity of this channel is required for the repolarization phase of the cardiac action potential; loss-of-function mutations cause long QT syndrome, a disease of prolonged cardiac repolarization predisposing to reentry arrhythmias. However, KCNQ1 is also expressed in other tissues, including pancreatic β-cells, where it regulates potassium current and thus, potentially, regulates insulin secretion. Single-nucleotide polymorphisms associated with increased KCNQ1 activity have been linked to susceptibility to type 2 diabetes mellitus due to reduced insulin secretion in several Asian populations. The converse has very recently been reported – fourteen individuals from six families with long QT syndrome who were discovered to have postprandial and symptomatic hyperinsulinemic hypoglycemia along with hypokalemia after oral glucose challenge. It is unknown how people with hypoglycemia would respond to medical therapy; however, the mechanism of the hypoglycemia suggests that they may be diazoxide unresponsive.
- 3.
AKT2 : AKT2 encodes one of three serine/threonine protein kinases that regulate a number of processes – cell proliferation, metabolism, survival, and angiogenesis. AKT2 is also involved in insulin signal processing downstream of the insulin receptor. Three known cases of hypoglycemia due to gain-of-function AKT2 mutations (heretozygous, meaning autosomal dominant, in two-thirds of cases, and mosaic in the remaining case) resulting in hypoglycemia have been reported. All three presented with severe hypoglycemia beginning at around 6 months of age. As the hypoglycemia in their cases was not caused by insulin but rather by a mediator of insulin’s effects, it was not amenable to treatment with diazoxide or octreotide; all required gastrostomy tube placement for frequent feeds to prevent hypoglycemia.
Summary
As the most common cause of hypoglycemia in infants and children, it is imperative that the diagnosis of HI be made swiftly in order to prevent further hypoglycemia and possible brain damage. Once the diagnosis has been made, the child should be started on dextrose-containing intravenous fluids while initiating therapy with diazoxide. Genetic testing will further aid in the management of the patient with HI, as the results can suggest possible focal disease, which can be cured with surgery, versus results that confirm diffuse disease. It is critical to understand the different genetic forms of HI in order to manage the patient with HI correctly. Lastly, for the surgical HI patient, it is crucial that they be cared for in a center that specializes in the management of HI and that has a team of endocrinologists, radiologists, pathologists, and surgeons that are all trained in the management of this disease.
Insulinomas
Insulinomas are insulin-producing islet-cell tumors. They are rare, with an estimated incidence of 1–4/1,000,000. While the majority of cases are sporadic, 10% are associated with a multiple endocrine neoplasia (MEN) type 1 syndrome. MEN1 results from inactivating mutations, deletions, or duplications in the MEN1 gene on chromosome 11q13.1, which encodes the tumor suppressor protein menin. Affected individuals present with episodic hypoketotic hyperinsulinemic hypoglycemia. MEN1 should be in the differential diagnosis for any insulinoma presenting in a child or young adult. Detectable insulin levels or a rise in blood sugar in response to glucagon at the time of hypoglycemia, along with other findings discussed in the previous section, establish excess insulin as the etiology for the hypoglycemia; a diagnostic fast of up to 72 h may be necessary to elicit the hypoglycemia. The vast majority of insulinomas are intrapancreatic; diagnostic imaging studies such as helical or multislice computerized tomography and magnetic resonance imaging (MRI) with gadolinium are good at identifying larger lesions, though endoscopic ultrasonography may be required to identify smaller lesions. Sequence analysis of 11q13.1 can identify mutations, deletions, or duplications in the MEN1 gene. Treatment of the insulinoma involves surgical excision in the 90% of cases in which the disease is localized to the pancreas; the hypoglycemia may also respond to diazoxide. Screening for other MEN1-associated neoplasias (pituitary tumors, parathyroid tumors, and other gastrointestinal neuroendocrine tumors) is a must in affected individuals.
Disorders of Glycogenolysis and Gluconeogenesis – “Glycogen Storage Diseases”
Disorders of glycogenolysis and gluconeogenesis, also called glycogen storage diseases (GSDs), are a group of disorders characterized by an inability to break down hepatic glycogen to form D -glucose or disorders in the hepatic process of gluconeogenesis – that is, converting noncarbohydrate precursors to D -glucose. As the liver does not have capacity to release free D -glucose, it no longer functions as a major player in glucose homeostasis. Depending upon the degree of impairment, the affected child is predisposed to hypoglycemia after varying durations of fasting. These disorders may also have a myriad of other features. In order to understand the GSDs, glycogenolysis and gluconeogenesis are reviewed briefly in the subsequent section.
Glycogen is a multibranched glucose polymer comprised of linear chains of glucose residues linked by α(1→4) glycosidic bonds. Branch points occur approximately every 10 residues, wherein a glucose residue on the stem chain is linked by α(1→6) glycosidic bonds to glucose on a new branch, which begins a new linear chain. Glycogen synthesis involves several steps, catalyzed by different enzymes. The liver is permeable to glucose; glucose is then converted into glucose-6-phosphate (G6P), preventing its exit, and then glucose-1-phosphate (G1P), which is the starting point for glycogen synthesis. In the initial step of glycogen synthesis, catalyzed by uridine-diphospate (UDP)-glucose pyrophosphorylase, G1P reacts with uridine-triphosphate (UTP) to form UDP-glucose. UDP-glucose monomers are then linked to each other via α(1→4) glycosidic bonds in a reaction catalyzed by glycogen synthase. The α(1→6) glycosidic bonds are created by the actions of glycogen branching enzyme. Glycogenolysis also entails the actions of several enzymes. Glycogen phosphorylase cleaves α(1→4) glycosidic bonds, yielding G1P and a linear glycogen chain shortened by one glucose residue; this does not work closer than four glucose residues to any branching point. Glycogen debranching enzyme transfers three of the four remaining glucose residues to the end of another glycogen branch, leaving only the branch-point glucose. Finally, the α(1→6) glycosidic bonds are cleaved by α(1→6) glucosidase, yielding a glucose molecule. G1P is converted by phosphoglucomutase to G6P, which in turn is hydrolyzed by G6P to yield free glucose and a phosphate residue. Finally (technically a step in gluconeogenesis rather than glycogenolysis), G6P is hydrolyzed to glucose via glucose-6-phosphatase (G6Pase). This step is crucial because phosphorylated glucose is charged and cannot easily cross the cell membrane to become available for use as energy in other tissues, while free glucose can easily diffuse across the cell membrane. The GSDs, or glycogenoses, are several inherited diseases due to abnormalities of the above-mentioned enzymes regulating the synthesis or degradation of glycogen. A deficiency in each of these enzymes is responsible for a different GSD, with variable impact upon fasting tolerance and various sequelae. The overall GSD incidence is estimated to be 1/20,000–1/43,000 live births. Glycogen is primarily stored in liver and muscle. Hypoglycemia is the primary manifestation of the hepatic glycogenoses; muscle glycogenoses are predominantly characterized by muscle weakness and muscle cramps with variable cardiac manifestations. Only the glycogenoses that cause hypoglycemia are discussed in this chapter; a detailed review of the muscle glycogenoses is beyond the scope of this review.
Gluconeogenesis is the metabolic pathway consisting of 11 enzyme-catalyzed reactions by which D -glucose is generated from noncarbohydrate substrates. Gluconeogenesis may begin in the mitochondria or cytoplasm, depending on the substrate. The primary amino acid substrates are alanine and glutamine, though others may contribute via the citric acid cycle. The other main gluconeogenic substrates are glycerol and lactate. In humans, the primary site of gluconeogenesis is the liver. The initial substrate for most, though not all, gluconeogenic reactions is pyruvate, which is generated from the aforementioned precursors (except glycerol) either by direct conversion of certain amino acids (alanine, cysteine, others), via the Cori cycle (lactate), α-ketoglutarate, and the citric acid cycle (glutamate and other amino acids), or via acetyl-CoA and the citric acid cycle (branched-chain amino acids). Many of the reactions are catalyzed by the same enzymes that catalyze the reversible steps in glycolysis; there are, however, four enzymatic steps that are unique to gluconeogenesis. The first is the mitochondrial carboxylation of pyruvate to oxaloacetate, catalyzed by pyruvate carboxylase (PC) using 1 ATP. Oxaloacetate is then reduced to malate using NADH, transported out of the mitochondrion into the cytosol, and oxidized back to oxaloacetate via NAD + . Oxaloacetate is then decarboxylated and phosphorylated to phosphoenolpyruvate (PEP) via the enzyme PEP-carboxykinase (PEP-CK), which is the second unique enzymatic step in gluconeogenesis is the decarboxylation and phosphate; one GTP molecule is hydrolyzed to GDP. Several steps then follow, which are the reverse of the glycolytic steps catalyzed by the same enzymes as in glycolysis; in one of these steps, glycerol (a byproduct of lipolysis) enters the gluconeogenic pathway. The third unique step is the conversion of fructose 1,6-bisphosphate to fructose-6-phosphate (F6P) by fructose 1,6-bisphosphatase (FBPase), releasing one phosphate molecule. This is the rate-limiting step in gluconeogenesis. One more reversible step follows, the conversion of F6P to G6P, then G6P is hydrolyzed to glucose via G6Pase, the final step of gluconeogenesis; as described earlier, this is where glucose released from glycogen enters the gluconeogenic pathway. Deficiencies in these enzymes are responsible for a different gluconeogenic disorder (generally considered in tandem with the GSDs), with variable impact upon fasting tolerance and various sequelae.
Disorders of Glycogenolysis
Table 3.2 provides a brief overview of GSDs; they are described in further detail in the subsequent section.
| GSD type | Eponym/other name | Gene/s | Affected protein/enzyme | Chromosomal location and inheritance * | Affected tissue | Diagnosis | Clinical features | Treatment |
|---|---|---|---|---|---|---|---|---|
| GSD Ia ** | Von Gierke’s disease | G6PC | Glucose-6- phosphatase, catalytic subunit | 17q21.31 AR |
| If undiagnosed/untreated
| Frequent carb-containing meals during the day
| |
| GSD Ib ** | Von Gierke’s disease | SLC37A4/G6PT1 | Glucose-6-phosphate transporter | 11q23.3 AR |
|
|
| |
| GSD IIIaGSD IIIb | Cori disease; glycogen debranching enzyme deficiency | AGL | Glycogen debranching enzyme | 1p21.2 AR | IIIa (80–85%)
| This enzyme has two independent catalytic activities, which occur at different sites on the protein: a 4-α-glucotransferase activity and an amylo-1,6-glucosidase activity If undiagnosed/untreated
| Similar to GSD I
| |
| GSD VI | Hers disease | PYGL | Glycogen phosphorylase | 14q21.1 AR | Liver | Sequencing of PYGL | Very rare except in Mennonites (affects 0.1% of Mennonite population) If undiagnosed/untreated
| |
| GSD IX 25% of all GSD cases | Fanconi–Bickel disease | PHK | Phosphorylase kinase – various isoforms | Varies | Varies | Sequence analysis of genes encoding affected subunits | Causes failure of hepatic glycogen phosphorylase activation Milder than GSD I or III If undiagnosed/untreated in early childhood:
|
|
| GSD IXa1, or GSD9A1 (formerly GSD 8) | Several subtypes | PHKA2 | Phosphorylase kinase α2 subunit – hepatic isoform | Xp22.13 X-linked |
|
| ||
| GSD IXb, or GSD9B | Most common form of GSD IX – 75% of all cases | PHKB | Phosphorylase kinase β subunit – hepatic and muscle isoform | 16q12.1 AR |
|
| ||
| GSD IXc, or GSD9C | PHKG2 | Phosphorylase kinase γ subunit – hepatic and testis isoform | 16p11.2 AR |
| Rare
| |||
| GSD 0 (or GSD 0a) | Glycogen synthase deficiency | GYS2 | Glycogen synthase 2 (hepatic) | 12p12.1 AR |
| Sequence analysis of GYS2 |
|
|
* See also http://www.genecards.org .
Glycogen Storage Disease Type “0”: Glycogen Synthase Deficiency
Glycogen synthase, as discussed earlier, catalyzes the rate-limiting step in glycogen synthesis in the liver and in skeletal muscle, namely, the transfer of glucose monomers from UDP-glucose to the terminal branch of the growing glycogen chain via the formation of α(1→4) glycosidic bonds. Several glycogen synthase isoforms exist – one specific to skeletal muscle (encoded by GYS1 ), and one specific to the liver (encoded by GYS2 ). Mutations in GYS2 cause glycogen synthase deficiency leading to GSD 0. Of note, the appellation of “glycogen storage disease” for GSD 0 is actually a misnomer; this is a disorder of glycogen formation rather than of aberrant glycogen breakdown.
In GSD 0, the lack of glycogen synthase activity leads to a decrease in hepatic glycogen content. Since excess dietary carbohydrates cannot be converted into glycogen, shunting into the glycolytic pathway and consequent lactate formation occur, resulting in postprandial lactic acid elevation, hyperglycemia, and sometimes postprandial glycosuria. However, once all dietary carbohydrate is consumed, gluconeogenesis and ketosis are triggered. Initially, gluconeogenesis blunts the impact of the hypoglycemia, but eventually, the ketosis and free fatty acid elevation due to lipolysis, fatty acid oxidation, and ketogenesis inhibit alanine release from skeletal muscle. Thus, the end result is postprandial ketotic hypoglycemia accompanied by elevated triglycerides and low alanine. To date, 15 different missense mutations in GYS2 leading to glycogen synthase deficiency have been reported to date; all are autosomal recessive in inheritance.
This disease may first manifest as infants transition to longer periods of overnight fasting in between feeds or may remain asymptomatic until a period of illness (e.g., gastroenteritis) leads to a prolonged fast, which is why some cases remain undiagnosed well outside of infancy and the toddler years. Fasting tolerance typically improves as children get older – adolescents may be able to fast as long as 18 h – but hyperketonemia is still frequently seen in the fasting state if the individual is untreated. Hypoglycemia can also occur even into adulthood in circumstances of prolonged fasting, pregnancy, gastrointestinal, or other illness leading to poor enteral carbohydrate intake, and upon strenuous exercise. Other manifestations in untreated individuals include short stature and osteopenia (frequently seen), cognitive delay in 22% of cases, and rarely seizures. These sequelae are preventable if hypoglycemia and hyperketonemia are prevented. Hepatomegaly is absent, as are other complications seen in other GSDs.
The diagnosis of GSD 0 is suggested by the presence of postprandial hyperglycemia, elevated lactates, and postprandial hyperketotic hypoglycemia on oral glucose tolerance testing. Definitive diagnosis of this condition can be done noninvasively via mutation analysis of the GYS2 gene. Liver biopsies are unnecessary.
Treatment of GSD 0 entails prevention of hypoglycemia by frequent daytime snacking (every two to four h) and, in young children, administering uncooked cornstarch (1–1.5 g/kg) at bedtime. Uncooked cornstarch should also be administered every 6 h during times of intercurrent illness and prior to strenuous or prolonged exercise. These interventions also have the effect of minimizing the ketosis and accumulation of free fatty acids to prevent hyperlipidemia and the suppression of skeletal muscle alanine release. Finally, affected individuals should consume a low-glycemic index diet with complex carbohydrates (as simple carbohydrates are more prone to trigger lactate elevation) and with protein supplementation to provide a greater quantity of gluconeogenic precursors in between meals.
Glycogen Storage Disease Type 1: Von Gierke’s Disease (Glucose-6-Phosphatase Deficiency)
G6Pase, as discussed earlier, catalyzes the final step of both glycogenolysis and gluconeogenesis in hepatocytes, intestinal mucosa, and renal epithelial cells – namely, the dephosphorylation of D -G6P to D -glucose and orthophosphate. G6Pase is a 35-kD endoplasmic reticulum (ER) integral membrane protein with nine transmembrane domains; the active domain faces into the ER. G6Pase is comprised of a catalytic subunit, which performs the dephosphorylation, and a transporter subunit encoded by SLC37A4 (chromosome 11q23.3), which handles the transport of G6P and of inorganic phosphate across the ER membrane. There are three G6Pase catalytic subunit-encoding genes in humans – G6PC , G6PC2 , and G6PC3 . Mutations in G6PC , a 12.5-kb single-copy gene comprised of five exons located on chromosome 17q21.31, are responsible for GSD 1a, while mutations in SLC37A4 are responsible for GSD 1b. GSD type 1 accounts for ∼25% of all GSDs diagnosed in the United States and Europe; overall prevalence is between 1/100,000 and 1/400,000 live births, with increased prevalence in some populations such as Ashkenazi Jews (prevalence 1/20,000). GSD 1a is far more common than 1b; 80% of GSD 1 patients have mutations in G6PC .
In both GSD 1a and 1b, hepatic production of glucose from both glycogenolysis and gluconeogenesis is impaired, because G6P cannot be converted to glucose. Since G6P cannot freely cross out of the liver and into the systemic circulation, the liver effectively cannot provide glucose to other tissues to use for energy. This results in severe hypoglycemia – as low as 40 mg/dL – within two to three h of feeding, as soon as the last bit of ingested carbohydrates has been metabolized. The hypoglycemia is accompanied by increased production of lactate, triglycerides, and uric acid. Onset of hypoglycemia typically occurs in infancy once the interval between feeding is lengthened, the infant starts feeding through the night, or an intercurrent illness interrupts the feeds. As glycogen synthesis is not impaired, glycogen accumulates in the liver, resulting in massive hepatomegaly and a protruberant abdomen as well as motor delay. Poor linear growth is frequently seen. Cognitive delay is infrequent, but may occur if recurrent hypoglycemia leads to cerebral damage.
GSD 1a
GSD 1a is far more common than 1b; 80% of GSD 1 patients have mutations in G6PC , with resulting absence of catalytic activity of G6Pase. GSD 1a is inherited in an autosomal recessive fashion, with either homozygous or compound heterozygous mutations; more than 80 different mutations have been described. The result of these mutations is an inability to generate glucose from carbohydrate or noncarbohydrate precursors, resulting in severe hypoglycemia and increased production of lactic acid, triglyceride, and uric acid, as above.
The hypoglycemia in GSD 1a is recurrent, seen multiple times daily in untreated patients, and is often asymptomatic due to the formation and utilization of other energy sources (e.g., lactic acid); symptomatic hypoglycemia is less common. In addition, because fructose and galactose must be converted to glucose via G6Pase prior to being available for use for energy systemically, their intake (and consequently intake of sucrose and lactose, which contain fructose and galactose, respectively) should be avoided or at least minimized in these patients – any ingestion does not aid systemic energy balance and only contributes to hepatomegaly and the laboratory abnormalities noted earlier. Thus, the diet for patients with GSD 1a and 1b is quite restrictive.
Metabolic manifestations of GSD 1a are as follows:
- •
Excess lactic acid production: This is the result of buildup of excess G6P in hepatocytes, with consequent buildup of glycolytic metabolites, exceeding the oxidative capacity of the citric acid cycle. The excess pyruvate gets shunted into production of lactic acid, a fuel the brain can utilize but which can contribute to metabolic acidosis.
- •
Hypertriglyceridemia: Some of the pyruvate is oxidated to acetyl-CoA in preparation for entry into the tricarboxylic acid (TCA) cycle; however, the levels of acetyl-CoA still exceed the capacity of the TCA cycle, and excess acetyl-CoA serves as substrate for cytosolic production of free fatty acids and eventually triglycerides, resulting in hypertriglyceridemia. Triglyceride elevations into the 400–800 mg/dL range and higher may be seen in poor metabolic control.
- •
Hyperuricemia: Excess G6P is produced, and some is shunted down the pentose phosphate pathway; this leads to increased production of ribose-5-phosphate and the subsequent de novo synthesis of the purine nucleotides. These excess purine nucleotides are subsequently degraded; this catabolic process produces uric acid. The uric acid also builds up due to decreased renal uric acid clearance in the presence of competition from lactate.
- •
Hyperalaninemia: Some excess pyruvate is also transaminated, forming alanine and resulting in hyperalaninemia.
Clinical manifestations of GSD 1a in untreated individuals include failure to thrive and consequent short stature (frequently seen), massive hepatomegaly and a protruberant abdomen (sometimes described as a Cushingoid appearance), “doll-like” facies in infants, motor developmental delay, metabolic acidosis (due to lactic acidosis) with consequent hyperventilation and apparent respiratory distress (infrequently seen), and hypoglycemic seizures (less commonly seen, as the brain can also utilize lactate as fuel). Social and cognitive development are only affected if the patient suffers cerebral damage from recurrent hypoglycemic seizures (uncommon). If metabolic control is not established and maintained, numerous complications may ensue, including:
- •
Metabolic acidosis: Impairment of gluconeogenesis results in chronic elevations of lactic acid (4–10 mM) even when the child is well. During intercurrent illness, prolonged fasting or during an episode of metabolic decompensation (which can mimic gastroenteritis), lactic acid levels can rise abruptly, sometimes exceeding 15 mM, and can produce severe metabolic acidosis. (The anion gap may be further increased by the production of uric acid, free fatty acids, and ketones). Symptoms include hyperpnea, emesis, and dehydration (all accompanied by hypoglycemia).
- •
Pancreatitis: May result from chronic hypertriglyceridemia. (Of note, pseudohyponatremia may also be seen, and serum may be lipemic.)
- •
Cardiovascular: Moderate elevations of phospholipids, total, and LDL cholesterol can be seen. The presence of these atherogenic lipoprotein profiles has been reported to be associated with the presence of cardiovascular disease risk measures, included carotid intima-media thickness, a measure of atherosclerosis, and increased central pressure wave augmentation, which predicts increased risk of cardiovascular events.
- •
Gout: This occurs due to chronic uric acid elevation; it is sometimes accompanied by nephrolithiasis. The nephrolithiasis also may have other etiologies – decreased citrate secretion in GSD 1a is seen with age; consequently, hypercalciuria develops, which can predispose to nephrocalcinosis and nephrolithiasis.
- •
Renal: Renal dysfunction may occur due to a combination of risk factors. Nephrolithiasis can develop as a complication of chronic uric acid elevation. In addition, proximal tubular dysfunction may be seen when metabolic control is poor, and includes glycosuria, phosphaturia, hypokalemia, and generalized aminoaciduria. These are reversible with improved metabolic control; however, chronic glomerular hyperfiltration is also seen in GSD 1a. In adulthood, long-term clinically silent hyperfiltration may lead to the development of chronic renal disease; this may include proteinuria, hypertension, and varying degrees of renal insufficiency. Renal biopsy in these cases may show focal glomerulosclerosis.
- •
Short stature and delayed puberty were frequently seen in patients with uncontrolled GSD 1 (a and b) in the past. Normal growth and pubertal development are typically seen in children who adhere to the specific GSD diet and consequently good metabolic control. However, GH secretory abnormalities and consequent lower GH levels may be seen even if linear growth is normal.
- •
Ovarian: Polycystic ovaries are frequently seen along with oligomenorrhea, thought to relate to insulin resistance; fertility may be preserved.
- •
Musculoskeletal: Decreased bone density is seen in over 50% of GSD 1a patients, and may relate to poor metabolic control as well as presence of other disease complications; decreased grip strength may also be seen.
- •
Platelet function: Abnormalities of platelet function are a common feature of GSD 1a, and include abnormal aggregation and low adhesiveness, decreased prothrombin consumption, and consequent prolonged bleeding time. These abnormalities correlate with degree of metabolic abnormalities and are reversible if good metabolic control is reached.
- •
Anemia: Anemia is frequently seen in GSD 1a. In preadolescents, iron-deficiency anemia is the predominant form, and may occur because uncooked cornstarch reduced the bioavailability of ingested iron. Over 50% of adults with GSD 1a may have anemia, of which anemia of chronic disease is the most common type. Severe anemia in GSD 1a adults may be associated with hepatocellular adenomas, most likely relating to the adenomas’ expression of hepcidin (which inhibits iron transport across gut mucosa); the anemia frequently improves following resection of the hepatic lesions.
- •
Liver: Hepatocellular adenomas, which predominantly develop during and after puberty, afflict up to 75% of patients with GSD 1a. Poor metabolic control, especially hypertriglyceridemia, may increase risk of developing the adenomas. Adenomas may be associated with chronic iron-resistant anemia. The adenomas can undergo malignant degeneration into hepatocellular carcinomas; hemorrhage may also occur.
The diagnosis of GSD 1a is suggested by the presence of severe recurrent hypoglycemia beginning three to four h postprandially, accompanied by the metabolic abnormalities listed earlier (elevated lactates, hypertriglyceridemia, and hyperuricemia), along with a rise in lactate in response to postprandial glucagon administration. On physical examination, massive hepatomegaly may be seen.
Diagnosis was formerly made via liver biopsy; GSD 1 (a and b) hepatic histopathologic findings include hepatocyte distension by glycogen and fat, uniform glycogen distribution, and nuclear hyperglycogenation. Large lipid vacuoles are frequently seen. However, definitive diagnosis of this condition can now be made noninvasively via mutation analysis of the G6PC gene. Liver biopsies are unnecessary.
The mainstay of therapy for GSD 1a is twofold – prevention of hypoglycemia and strict adherence to a specialized diet:
- •
Hypoglycemia prevention: In infants (<6–12 months of age), this can be accomplished with frequent meals or snacks (every two to four h) during the daytime and by continuous infusions of dextrose via gastrostomy tubes overnight. In older infants and children, similar results can be obtained by administering uncooked cornstarch (1–1.5 g/kg, dissolved in formula, water or sweetened beverages such as Kool-Aid) every three to five h during the day and every four to six h overnight.
- •
Other dietary interventions: Carbohydrates typically provide 60–65% of calories. In order to minimize hypertriglyceridemia, dietary fat is restricted to no more than 20% of total energy intake, and cholesterol intake is restricted to <300 mg/day. Foods that contain fructose and/or galactose (including sucrose and lactose containing foods) must be restricted, in order to minimize intake substrates that require action of G6Pase in order to be available for systemic use.
- •
When hypoglycemia and hyperlacticacidemia are prevented, hepatic size decreases, linear growth improves, and serum uric acid and lipids (especially triglycerides) normalize. Allopurinol may be used as adjunct therapy if severe hyperuricemia persists despite good metabolic control. Therapy with triglyceride-lowering agents (e.g., gemfibrozil) is seldom required. Serum alpha-fetoprotein and hepatic ultrasound are performed annually to screen for hepatic adenomas.
GSD 1b
GSD 1b is far less common than 1a, comprising only 20% of cases of GSD 1. GSD 1b results from a mutation in SLC37A4, the gene-encoding G6P transporter 1 ( G6PT1 ), which regulates the rate-limiting step of G6P transport through the ER membrane and also plays a role in calcium sequestration in the ER lumen. GSD 1b is also inherited in an autosomal recessive fashion, with either homozygous or compound heterozygous mutations; more than 35 different mutations have been described. The result of these mutations is inability to generate glucose from carbohydrate or noncarbohydrate precursors, resulting in severe hypoglycemia and increased production of lactic acid, triglyceride, and uric acid, as in GSD 1a.
The hypoglycemia in GSD 1b is very like that seen in GSD 1a – recurrent, seen multiple times daily in untreated patients, and often asymptomatic. The other clinical features of GSD 1b and the long-term complications are very similar to those of GSD 1a. However, GSD 1b carries with it a few additional comorbidities. Most GSD 1b patients suffer from neutropenia, either cyclic or constant, accompanied by neutrophil and monocyte dysfunction, which results in an impaired respiratory burst, defective bactericidal activity, and increased susceptibility to infection, especially skin and pulmonary infections. The neutropenia is thought to result from enhanced ER stress as a consequence of loss of G6PT activity; this in turns leads to increased oxidative stress and apoptosis. In addition, GSD 1b patients may also display symptoms of inflammatory bowel disease, including oral aphthous ulcers, perianal abscess or fistulas, and colitis, which may be indistinguishable from Crohn’s disease. Finally, an increased risk of autoimmune disorders – including thyroiditis, GH deficiency, and myasthenia gravis – in GSD 1b has occasionally been reported.
The diagnosis of GSD 1b, as in 1a, is suggested by the presence of severe recurrent hypoglycemia beginning 3–4 h postprandially, accompanied by the metabolic abnormalities listed earlier (elevated lactates, significant hypertriglyceridemia, and hyperuricemia) and a rise in lactate in response to postprandial glucagon administration. On physical examination, massive hepatomegaly may be seen, along with short stature and failure to thrive. The diagnosis of GSD 1b rather than 1a is suggested by the presence of neutropenia. Histopathological findings are similar to those seen in GSD 1a; however, definitive diagnosis of this condition can be done noninvasively via mutation analysis of the SLC37A4 gene, rendering liver biopsies unnecessary.
The mainstay of treatment of GSD 1b is very similar to that of GSD 1a: it entails adherence to a fructose- and galactose-free diet, frequent feeds (every 2–4 h) during the day with overnight gastrostromy tube feeds or dextrose in infants, and uncooked cornstarch 1–1.5 g/kg every 3–5 h during the day or every 4–6 h at night. Liver transplantation may be indicated for those with hepatic malignancy. Granulocyte colony stimulating factor (G-CSF) has been widely used since the 1990s to treat the chronic neutropenia associated with GSD 1b; however, caution must be utilized, as there have been case reports of acute myelogenous leukemia developing as a consequence of G-CSF therapy.
GSD III (Cori Disease) – Glycogen Debrancher Enzyme Deficiency
As discussed earlier, glycogenolysis entails the actions of several different enzymes. The major one is glycogen phosphorylase, which cleaves α(1→4) glycosidic bonds between adjacent glucose moieties in glycogen. However, 1 in 10 glucose residues in glycogen are branch points, existing as α(1→6 linkages). As glycogen phosphorylase stops cleaving when it reaches a point four residues from a branching point, the actions of glycogen phosphorylase alone do not suffice to mobilize glycogen stores. To mobilize the rest, the actions of another enzyme are required – glycogen debranching enzyme, encoded by the AGL gene and located on chromosome 1p21.2. The AGL gene is 85 kbp long and is composed of 35 exons. Six major mRNA isoforms have been identified. All isoforms share the same translation initiation site in exon 3, but differ in the 5′-untranslated regions due to differential transcription relating to specific cryptic splice sites located upstream from the translation initiation site and which enable exon removal or retention. Isoform 1, the major isoform, contains exon 1 and lacks exon 2; it is the most widely expressed, and is found in liver, kidney, and lymphoblastoid cells, as well as in myocytes; it encodes a 1532-amino acid residue protein. Isoforms 2–4 lack exon 1 and are exclusively expressed in cardiac and skeletal myocytes. All major isoforms contain exons 4–35.
Debranching enzyme is a 165-kD protein that acts as a 1,4-alpha- d -glucan:1,4-alpha- d -glucan 4-alpha- d – glycosyltransferase and amylo-1,6-glucosidase in glycogen. The enzyme has two different, independent catalytic activities that take place at different sites and can function independently of each other – 4-alpha-glucotransferase activity and amylo-1,6-glucosidase activity. GSD III results from an autosomal recessive mutation in AGL, due either to homozygous or to compound heterozygous mutations or deletions. Missense, splice site and nonsense mutations, small frameshift deletions and insertions, and large gene deletions and duplications have all been described in the AGL gene. The majority of GSD III patients have deficient enzyme activity in both liver and muscle (GSD IIIa), while ∼15% have insufficient enzyme activity in the liver only (GSD IIIb). GSD IIIa mutations possess considerable allelic heterogeneity; GSD IIIb, on the other hand, is primarily associated with two mutations in exon 3 of AGL – c.18_19delGA (p.Gln6HisfsX20), formerly described as c.17_18delAG, and c.16C > T (p.Gln6X). Most patients with GSD III lack enzymatic activity at both catalytic sites (both the 4-α-glucotransferase and the amylo-1,6-glucosidase sites); deficiencies only of the glucosidase activity (GSD IIIc) or of the glucotransferase activity (GSD IIId) have been reported, but are rare. The incidence of GSD III in the US population is 1/100,000, although it is more common in certain subpopulations, such as individuals of North African Jewish descent, where the prevalence is estimated to be 1/5400.
Hypoglycemia in GSD III occurs due to an inability to fully utilize glycogen. Glycogen debrancher enzyme (GDE) deficiency leads to accumulation of abnormal glycogen possessing short outer chains in the liver, phosphorylase limit dextrin, in affected tissues. It is typically seen after more prolonged fasts than in GSD I, is accompanied by ketosis, and is usually milder than in GSD I because gluconeogenesis is not impacted, and hepatic glycogen can be metabolized to a certain extent via glycogen phosphorylase. However, some patients with GSD III have hypoglycemia equal in severity to patients with GSD I. Other biochemical abnormalities include hyperlipidemia and transaminase elevation of over 500 U/L are often seen (the latter primarily in childhood). Uric acid and lactate concentrations are typically normal, in contrast to GSD 1a and 1b. Profound ketosis can develop after overnight fast.
The clinical phenotype of GSD III is somewhat variable. However, the main underlying clinical features in childhood are hepatomegaly, fasting hypoglycemia as above, and hyperlipidemia; if treatment is not initiated, growth failure and pubertal delay can result. Renal disease is not typically seen. In GSD IIIa, where skeletal and/or cardiac muscle is involved, a vacuolar myopathy may develop, and serum creatine kinase (CK) elevations of 5–45x above the upper limit of normal may be seen. Long-term complications of GSD III can include:
- •
Hepatic cirrhosis: The hepatomegaly associated with GSD III usually resolves postpuberty, which may be attributable to a combination of lower glucose requirements but also to possible development of livery cirrhosis, which can progress to end-stage liver disease.
- •
Hepatic adenomas, whose prevalence in GSD III is estimated at between 4% and 25%, and rarely hepatocellular carcinomas.
- •
Skeletal myopathy: Motor delays may be the initial signs, followed by slowly developing weakness and wasting of both proximal and distal muscles. Nerve conduction studies may be abnormal. Muscles involved in respiration are typically spared. Skeletal myopathy may occur in isolation, or in combination with cardiomyopathy.
- •
Osteoporosis: Seen primarily in GSD III patients with skeletal myopathy.
- •
In women, polycystic ovaries may develop at higher rates; however, fertility is not typically impacted.
- •
Cardiomyopathy: Ventricular hypertrophy is commonly seen; overt cardiac dysfunction is rare, but congestive heart failure may occasionally develop.
- •
Sudden death: Rarely seen; thought to be secondary to cardiomyopathy-related arrhythmias.
The diagnosis of GSD III is suggested by fasting hypoglycemia accompanied by ketosis in the setting of hepatomegaly and hyperlipidemia, especially if CK elevation and/or transaminitis is seen. The absence of uric acid and lactate elevation help to differentiate from GSD I (though uric acid elevations may be seen following exertion) muscle. Glucagon administration after an overnight fast does not trigger a rise in blood glucose levels, but if glucagon is administered 2 h following a carbohydrate-rich meal, blood glucose levels do increase normally, again in contrast to GSD I. In most GSD III individuals, liver biopsy (or muscle biopsy in those patients with myopathy) shows cytosolic vacuolar accumulation of nonmembrane-bound glycogen. The excess glycogen that accumulates is structurally abnormal (short outer branches); however, lipid vacuoles are less common than in GSD I. Hepatic fibrosis ranging in severity from periportal fibrosis to micronodular cirrhosis may be seen. Diagnosis of GSD III may be based on demonstration of deficient GDE activity in biopsied hepatocytes and/or myocytes. However, biopsy is no longer necessary to establish a diagnosis; molecular genetic testing for pathogenic AGL mutations on both alleles is sufficient to establish the diagnosis noninvasively .
Treatment of GSD III entails avoidance of prolonged fasting, typically involving uncooked cornstarch (1.75 g/kg), though at longer intervals than in GSD I – every 6 h. This avoids hypoglycemia, improves linear growth, and reduces the transaminitis. For patients too young for cornstarch or who have significant growth failure and/or myopathy, continuous tube feeds overnight of a mixture of glucose and protein (or glucose oligosaccharides and amino acids) is beneficial. There is no need to restrict fructose or galactose in GSD III as in GSD I, as gluconeogenesis is intact. Serum alpha-fetoprotein and hepatic ultrasound are performed annually to biannually to screen for hepatic adenomas; liver transplants are performed, but rarely. Patients with skeletal myopathies should have intermittent electrocardiograms and echocardiograms. The prognosis for patients with myopathy is less favorable than for those with isolated hepatic involvement. Physical therapy and aerobic training may be beneficial for patients with myopathies.
Glycogen Storage Disease VI (Hers Disease, Glycogen Phosphorylase Deficiency) and GSD IX (Phosphorylase Kinase Deficiency)
In the liver, glycogen phosphorylase cleaves α(1→4) glycosidic bonds, yielding G1P and a linear glycogen chain shortened by one glucose residue. Glycogen phosphorylase has two configurations – the inactive configuration, phosphorylase B, and the active configuration, phosphorylase A. The conformational shift to the more active configuration is triggered by phosphorylation of two serine residues by phosphorylase kinase (PhK). Glycogenoses caused by reduced glycogen phosphorylase activity (GSD VI and IX) are a heterogeneous group, clinically indistinguishable but of different etiologies; some result from loss of function of glycogen phosphorylase itself (GSD VI), while others are due to hepatic phosphorylase kinase deficiency (GSD IX) resulting from mutations in one of the genes encoding PhK subunits.
Glycogen phosphorylase has three different isoforms in humans encoded by three different genes; the isoforms are respectively expressed primarily in the liver, brain, and muscle. The hepatic isoform is encoded by the PYGL gene, located on chromosome 14q22.1; the classic form of GSD VI results from loss-of-function mutations in or deletions of PYGL (homozygous or compound heterozygous mutations, deletions or duplications of one or more exons or whole-gene deletions or duplications; at least 12 different pathogenic mutations are known). Glycogen phosphorylase mutations are autosomally recessively inherited, and the deficiency is relatively rare except in the Mennonite population, where they are relatively common due to a founder mutation, affecting 0.1% of the population.
PhK is a serine-threonine phosphorylase comprised of a homotetramer of tetramers, for a total of 16 subunits; each of the four tetramers is composed of four different subunits, each encoded by a different gene: α (encoded by PHKA1 and PHKA2 ), β (encoded by PHKB ), γ (encoded by different PHKG genes for different tissues – the hepatic/testis-specific isoform is encoded by PHKG2 ), and δ (encoded by CALM1 ). The enzyme’s catalytic activity occurs at the γ subunit; the α and β subunits have regulatory functions; the δ subunit, a calmodulin, mediates the calcium dependence of the enzyme. PhK is encoded by several different genes:
- •
PHKA1 , located on chromosome Xq13.2, encodes a muscle isoform of subunit α; mutations therein result in X-linked muscle PhK deficiency, a rare X-linked disorder.
- •
PHKA2 , located on chromosome Xp22.13, also encodes a hepatic isoform of subunit α; PHKA2 mutations cause hepatic PhK deficiency, or GSD9A, also known as X-linked liver glycogenosis (XLG). XLG is the most common variant of GSD IX, responsible for ∼75% of cases of GSD IX. These mutations are subdivided into several categories, including GSD IXa1, where there is no PhK activity in hepatocytes or erythrocytes, and GSD IXa2, where the PhK activity is diminished or absent in hepatocytes but preserved in erythrocytes. At least 28 different PHKA2 mutations have been identified. Both GSD9A1 and GSD9A2 are caused by mutations in PHKA2 ; however, GSD9A2 is caused by either small in-frame deletions or insertions or by missense mutations, explaining the residual enzymatic expression in erythrocytes.
- •
PHKB , located on chromosome 16q12.1, encodes subunit β in liver and muscle; mutations, which are rare, cause autosomal recessive GSD9B. Fourteen different pathogenic variants have been described, including missense and nonsense mutations, splice site variations, and small insertions or deletions.
- •
PHKG2 , located on chromosome 16p11.2, encodes the catalytic δ subunit of PhK in the liver and testis. Mutations cause autosomal recessive hepatic PhK deficiency, GSD9C. Nine different pathogenic mutations have been described.
Phosphorylase kinase mutations are considerably more common than glycogen phosphorylase mutations, and are collectively responsible for ∼25% of all GSDs, with an estimated prevalence of 1/100,000. GSD9A, 9B, and 9C are clinically indistinguishable from each other, as well as from GSD XI.
GSD VI and XI have milder phenotypes than GSD I or III and a more favorable prognosis. Symptomatic hypoglycemia is uncommon, and primarily occurs after prolonged fasting or intensive exercise; it is accompanied by ketosis. Relatively mild hyperlipidemia (triglycerides and other lipids) and mild transaminase elevation may be seen; lactic acid and uric acid are typically normal, and thus metabolic acidosis is uncommon. Affected individuals usually present in infancy or early childhood due to growth retardation and hepatomegaly. If muscle as well as hepatic glycogen phosphorylase is impacted ( PHKA1 , PHKB ), hypotonia and motor delay may be present. As patients age, the laboratory abnormalities and hepatomegaly typically improve, as does linear growth – often without treatment. Adults are often asymptomatic. Long-term complications have been reported – liver cirrhosis, CNS complications such as impaired cognition, speech impairment, seizures, peripheral sensory neuropathy, and/or renal impairments such as tubular dysfunction and proximal renal tubular acidosis. However, these long-term complications are all rare.
Diagnosis of GSD VI can be made by assaying phosphorylase activity in erythrocytes. PHKB activity can also be assessed in erythrocytes or leukocytes, but normal PhK activity in blood does not definitively rule out GSD9, given the possibility of XLG due to PHKA2 . Hepatic biopsies for enzyme activity are possible, but again unnecessary; molecular genetic diagnosis of mutations or deletions in PYGL , PHKA1 , PHKA2 , PHKB , or PHKG2 is the test of choice.
GSD VI and IX are easier to treat than GSD I or III. No special diet is required, and there is no need to avoid fructose- or galactose-containing foods. Prolonged fasting should be avoided, and plenty of carbohydrates should be ingested if strenuous exercise is undertaken. Bedtime snacks often suffice to prevent nocturnal hypoglycemia and ketosis; if not, 2 g/kg of uncooked cornstarch can also be administered at bedtime. Overnight feeds are not required.
Disorders of Gluconeogenesis
Gluconeogenic disorders are summarized in Table 3.3 .
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


