74 Hyperthermia
Hyperthermia (HT) is the use of elevated temperature for the treatment of cancer, in this case, typically using temperatures in the range of 41° C to 45° C for 1 hour or more. Tumor regression after a high fever caused by erysipelas was first reported in the medical literature in 1866 by the German physician W. Busch.1 This case and others led a New York surgeon, W. B. Coley, to treat cancer patients with bacterial pyrogens (Coley toxin).2 In 1910, Müller described the potential of HT as an adjuvant to radiotherapy (RT).3 However the biologic rationale for applying HT with RT in cancer therapy was not investigated in depth until the 1970s.4–6 Exciting laboratory studies demonstrating the activity of heat against tumor cells in tissue culture and animal models encouraged numerous nonrandomized clinical trials of RT plus HT in small superficial tumors.7 Promising results of these early trials led to premature randomized trials, before adequate thermometry or hyperthermic delivery systems were available, with disappointing results.8,9 Since then, multiple prospective, randomized trials have shown a benefit of adjuvant HT,10–15 and thermal dose-response analyses have predicted appropriate thermal dose prescriptions for superficial and deep tumors.16 New protocols that prescribe higher minimum thermal doses or combine HT simultaneously with RT for increased thermal radiosensitization are nearing completion. Encouraging trials of HT combined with chemotherapy or liposomal chemotherapy have been performed. There is also recent interest in using HT as a modulator for gene therapy or immunotherapy. However, for HT to become an established treatment modality, much work is needed to (1) better understand and exploit the biology of heat in combination with RT or chemotherapy or novel therapies; (2) improve the equipment used for performing, monitoring, and planning hyperthermic treatments; and (3) further define appropriate thermal dose goals and clinical applications. This chapter reviews the biologic rationale for HT, physical principles of techniques used to heat tissues, clinical results, and future directions of HT research.
Biologic Rationale
Heat kills cells as a function of time and temperature and also sensitizes cells to radiation damage. Evidence suggests that protein denaturation is involved in this process, whereas direct damage to deoxyribonucleic acid (DNA) is not.17 Although the exact mechanisms of hyperthermic cell killing are still not clear, much progress has been made in the past decade in understanding the molecular, biochemical, and cellular consequences of thermal stress. Heating induces a variety of intracellular changes: activation of heat shock transcription factors; enhanced synthesis of heat shock proteins (HSPs); and alterations in nuclear and cytoskeletal structures, cellular metabolism, macromolecular synthesis, intracellular signal transduction, and hormone-receptor interactions.18
Cellular Response to Heat
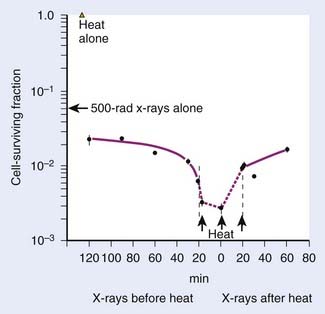
Exposure of mammalian cells to temperatures higher than 40° C leads to reproductive cell death. This effect of hyperthermic treatment on cells’ reproductive capacity depends on both the applied temperature and the duration of the exposure, and can be represented as survival curves resembling x-ray survival curves (Fig. 74-1). When the surviving cell fraction (capable of reproduction) is plotted on a logarithmic scale versus the duration of heating on a linear scale, the survival curve is characterized by an initial shoulder region followed by an exponential decrease in the surviving fraction.5 The time required to reduce the survival in the exponential region to 37% of its initial value is defined as D0.
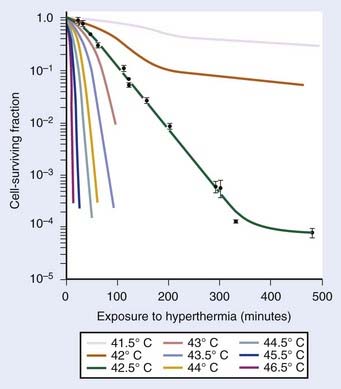
FIGURE 74-1 • Survival curves for asynchronous Chinese hamster ovary cells heated at different temperatures for varying time.
(From Hall EJ: Radiobiology for the radiologist. Philadelphia, 1994, JB Lippincott Company, p 260.)
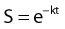
in which S is survival, t is the treatment time, and k is the inactivation rate at the treatment temperature.19 The term k may also be replaced by 1/D0 as used in RT biology.6 This model does not account for the survival curve “shoulder.”
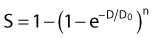
in which n is the number of targets to be inactivated before the cell is killed.6,20 The survival curves for this model are characterized by the parameters n, D0, and Dq (the quasithreshold dose) in which Dq = (ln n)D0. Using this model, one can construct Arrhenius plots to demonstrate the kinetics of heat killing, where 1/D0 is plotted against the inverse of the absolute temperature (Fig. 74-2). For most cell lines studied, the Arrhenius plots of cell inactivation seem to be composed of two segments, with a break at or near 43° C.4 Above the break point, the activation energy is between 110 and 150 kcal/mol, which is the energy required for protein denaturation,21 suggesting that protein damage is mainly responsible for cell death caused by HT. At lower temperatures, the activation energy is higher, approximately 300 to 400 kcal/mol. On the basis of the difference in activation energies, it has been suggested that there may be different primary modes of heat-induced cell death, one dominant higher than 43° C and the other lower than 43° C. However, the change in the slope of Arrhenius plots lower than 43° C could simply be a manifestation of the ability of the cells to develop thermotolerance, the transient resistance of cells to heat killing.22 The Arrhenius plot for a given cell line can be modulated by thermotolerance or environmental parameters such as pH.
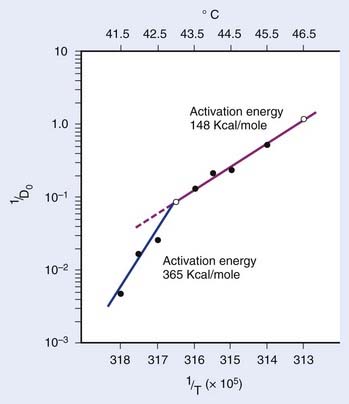
Using the activation enthalpy and entropy for hyperthermic cell killing (determined from Arrhenius plots based on D0s), Lepock and colleagues23 introduced a thermodynamic model for hyperthermic cell killing based on the existence of a critical target. The rate of cell killing is thought to correspond with the rate of inactivation and denaturation of this hypothetical critical target. The calculated transition temperature of this critical target appeared to be 46.0° C for V79-WNRE cells when the temperature was raised by 1° C per minute. When the scan rates of 0.1° C per minute or 10° C per minute were used for differential scanning calorimetry (DSC), the calculated transition temperature was 43.5° C or 48.5° C, respectively. Thus, transition temperatures are only meaningful when the scan rate used in the measurements is specified.23 Cellular fractionation to localize the critical target showed that protein denaturation occurred at 46° C in all subcellular fractions.
A fourth model, which nicely describes heat killing under various conditions (single heating, thermotolerance, and step-down heating), was proposed by Jung.24 This model assumes a two-step process. The first step is the production of nonlethal lesions that can be converted into lethal lesions upon further heating in the second step. After the cells are heated for a time t at a certain temperature, the surviving fraction S is given by the equation:
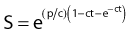
in which p is the rate constant for the production of nonlethal lesions per cell per unit of time and c is the rate constant for the conversion of a nonlethal lesion into a lethal lesion per unit of time. One lethal lesion is sufficient to kill a cell. Jung assumed that the production and conversion of nonlethal lesions were random and the rate constants depended only on the temperature of the heat treatment.
The intrinsic thermal sensitivity of different cell lines varies significantly. So far, no consistent difference has been demonstrated between normal and malignant cells. On the other hand, the extracellular milieu can modify cells’ thermal sensitivity. In general, cells that are at low pH or are nutrient-deprived are more sensitive to heat. These cells may be hypoxic cells that are three times more resistant to radiation than are normally oxygenated cells. However, cells exposed to low pH for long periods adapt to pH changes and lose their increased heat-sensitivity.25,26 Sensitivity to heat shock as a function of cell cycle has also been extensively studied. It is well established that the age response of thermal sensitivity complements that for x-irradiation. The phase of the cell cycle most resistant to x-rays (late S phase), is most sensitive to heat shock response.27
When more than one heat treatment is given, cellular thermal sensitivity to the combined treatments can be modified depending on the order of the heat shock temperatures involved. Step-up heating occurs when the second heat shock is at a higher temperature than the first heat exposure at 39° C to 42° C. Conversely, step-down heating occurs when the first treatment is a short exposure at a high temperature, greater than 43° C, immediately followed by a subsequent exposure to less than 43° C. During step-up heating, thermotolerance may be induced if the first treatment temperature is less than 43° C. Therefore, the cytotoxic effect of combined heat treatments is much less than the effect without preheat treatment. On the other hand, during step-down heating, preheat treatment at greater than 43° C for a short period can sensitize cells to a subsequent heat shock treatment at a lower temperature, which may have been nonlethal by itself without the preheat treatment. However, a high temperature pulse does not abolish any thermotolerance developed earlier.6
Thermal Dose
Because heat killing is a function of both time and temperature, a thermal dose unit is needed to combine both factors and account for the different effect of heat higher and lower than 43° C. Equivalent minutes at 43° C (EM 43° degrees) was proposed as the thermal dose unit by Sapareto and Dewey in 1984:28
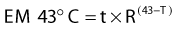
in which t is the time at temperature T, the constant R = 0.5 for temperatures higher than 43° C, and R is 0.25 less than 43° C. Thermal dose for a given treatment can be obtained by summing thermal doses for each minute of treatment, and the thermal dose from multiple treatments can be summed to obtain the cumulative equivalent minutes (CEM) at 43° C for an entire treatment course, assuming that the interval between heat treatments is long enough to allow for decay of thermotolerance. To describe the variations in thermal dose throughout a tumor, minimum, maximum, and average values can be given as well as thermal dose values for the T90 and T50 temperatures (the temperatures exceeded by 90% or 50% of the measured intratumoral points, respectively), and so on.
This concept of thermal dose has been validated in multiple pet animal and human clinical trials. Multivariate analysis of a randomized trial in pet animals with spontaneous tumors treated with RT alone or RT plus HT showed that, along with tumor volume, the non–site-specific average minimum EM 43° was the most important predictor of complete response (CR) (P < 0.001) and local control (P < 0.05). The CR rate was only 32% for 28 tumors with an average minimum CEM 43° (CEM 43° Tmin) of 1.0 minute or less, but 68% for 79 tumors with greater than 1.0 average CEM 43° Tmin.29 A thermal dose analysis of a prospective, randomized trial in patients with recurrent breast cancer showed a significant association between CEM 43° Tmin and response, with CR rates of 43% for CEM 43° Tmin less than or equal to 10 minutes versus 77% for CEM 43° Tmin greater than 10 minutes.30 Retrospective analyses of HT patients treated at Duke University showed that the cumulative minutes of treatment for which T90 exceeded 39.5° C was highly associated with CR of superficial tumors,31 and that the cumulative minutes of treatment for which T50 exceeded 41.5° C was highly associated with greater than 80% necrosis of soft-tissue sarcomas treated preoperatively with thermoradiotherapy.32 Thermal dose-response formulas based on these data showed that T90 and T50 temperatures routinely achieved need to be improved by 1.2° C to 1.5° C, or treatment duration increased threefold to fivefold, to increase thermal dose sufficiently to justify phase III trials.16 There were also significant associations between CEM 43° T90 and response in a study of preoperative regional HT combined with conventional RT and low-dose 5-fluorouracil chemotherapy and leucovorin for 37 locally advanced primary rectal cancers (P = 0.006)33 and between CEM 43° T90 and CR and local control in patients with recurrent breast adenocarcinoma treated at Stanford with RT and HT.34 These parameters, CEM 43° Tmin and CEM 43° T90, have become standard thermal dose descriptors for superficial HT.
Mechanisms of Action of Heat
Unlike ionizing radiation, HT does not result in the localized deposition of high levels of energy in cells. The thermal energy is more or less evenly absorbed by all molecules in the cell. As mentioned previously, the activation energy obtained for cultured cell lines and transplantable tumors in the temperature range from 42.5° C to 47° C is approximately 150 kcal/mol,6,27 suggesting that protein denaturation is the main cause of hyperthermic cell killing.
More direct evidence that protein denaturation is responsible for thermal killing was provided using DSC.23 In eukaryotes, the onset for protein denaturation was found to be approximately 40° C. Thermotolerance, cycloheximide, and D2O all increased the thermostability of proteins and also resulted in increased survival levels after HT.35 These results were further confirmed by studies conducted by Burgman and colleagues36 using electron spin resonance measurements and thermal gel analysis, showing that a correlation existed between protein denaturation and heat killing.
Protein denaturation and subsequent aggregation appear to occur throughout the entire cell, including cytoplasmic, nuclear, and membrane proteins.36 It is unlikely that a single type of protein is the critical target for cell killing by heat. All subcellular structures contain temperature-sensitive proteins that denature and may subsequently aggregate.37 Small changes in temperature can drastically alter the structure of plasma membranes and impair many membrane-related functions that, by themselves or in combination with other protein damage in the cell, could lead to cell death.38,39 Heat has also been shown to alter the mitotic spindle and centrosome organization, resulting in the formation of multinucleated, nonclonogenic cells,40 and to damage mitochondria,41 with heat-induced inhibition of metabolic functions such as glycolysis and respiration. Heat damages polysomes and microsomes and inhibits the synthesis of proteins, ribonucleic acid (RNA), and DNA in a thermal dose–dependent manner.39,41 The inhibition of RNA and DNA synthesis appears to be due to heat-induced changes in chromatin structure caused by the denaturation and aggregation of nuclear proteins. Heat-induced changes in the nuclei of eukaryotic cells include the appearance of actin bundles; increased vesiculation; the reduction of intact nucleoli41; and a dramatic, dose-dependent increase in protein content in isolated chromatin, nuclei, nuclear matrices, and nucleoids.35,42,43 This appears to be at least partially due to decreased leakage of proteins such as DNA and RNA polymerases from the nucleus during isolation as the heat-denatured proteins become aggregated and thus insoluble. This phenomenon is referred to as heat-induced nuclear protein aggregation. It is likely that nuclear protein aggregation, which occurs largely at the nuclear matrix, interferes with DNA replication, transcription complex interactions, and DNA unwinding needed for DNA replication and DNA repair.44
Interestingly, on exposure to heat, a group of proteins called heat shock proteins (HSPs) translocate from the cytoplasm to the nucleus.41,44,45 It has been shown that HSPs protect against protein aggregation or facilitate disaggregation of heat-induced insoluble protein complexes in cell lines and yeast.46–48
Thermotolerance and Heat Shock Proteins
One of the most interesting aspects of thermal biology in the mammalian system is the response of heated cells to subsequent heat challenges. Mammalian cells, when exposed to a nonlethal heat shock, have the ability to acquire a transient and nonheritable resistance to subsequent exposures at elevated temperature. This phenomenon, first shown by Henle and Leeper49 and Gerner and Schneider,50 has been described as induced thermal resistance, thermal tolerance, or, most commonly, thermotolerance. Several excellent reviews have discussed this phenomenon in considerable detail.51–53
In vitro, acute thermotolerance can be induced by a short initial heat treatment at temperatures higher than 43° C followed by a 37° C incubation before the second heat challenge. Thermotolerance can also be induced during a continuous and long exposure (1 to 20 hours) at temperatures lower than 43° C (chronic thermotolerance).54–56 The degree of thermotolerance developed can be dramatic; increases in survival levels by several orders of magnitude are commonplace. The thermal history, heat fractionation interval, and recovery conditions all significantly modify the kinetics of thermotolerance.52,53
Thermotolerance can also be induced by treatment of cells with a variety of chemicals followed by a drug-free period before the heat challenge. Some examples of the thermotolerance-inducing chemicals are heavy metals, ethanol, sodium arsenite, procaine, lidocaine, aliphatic alcohols (C5-C8), dinitrophenol, carbonyl cyanide m-chlorophenyl hydrazone, puromycin, and prostaglandins.56–58 Pretreatment with heat or chemicals induces both thermotolerance and resistance against the cytotoxicity of the tolerance-inducing chemicals. This development of cross-resistance suggests overlap in the mechanisms of induction of tolerance by heat and chemicals.
The amount of thermotolerance expressed depends on the temperature and duration of the first heat treatment and the interval between the two heat treatments (Fig. 74-3).59 At temperatures of 43° C or higher, thermotolerance does not develop during the first heat exposure; a subsequent incubation at 37° C is required for its expression. In contrast, for initial treatment at 41° C, thermotolerance is almost completely developed at the end of the first heat treatment. Based on these data, an operational model for the development of thermotolerance was formulated with three phases: an initial event (trigger), the expression of resistance (development), and the gradual disappearance of resistance (decay).59 First, the triggering event converts normal cells to the triggered state with a rate constant k1. This process very likely involves the activation of the heat shock transcription factor-1 (HSF1)60,61 and probably an additional regulatory factor or factors.62 Second, these triggered cells are converted to thermotolerant cells with a rate constant k2. Higher than 43° C, k2 = 0; the triggered cells remain sensitive, and only after being transferred to 37° C do they convert to their thermotolerant state. This thermotolerant state, in general, is associated with the elevated expression of HSPs, and enhanced protection against and faster recovery from thermal damage. Finally, thermotolerant cells all reconvert to their sensitive state at a slower rate governed by rate constant k3.
The HSPs are usually identified by their molecular mass; for example, HSP70 is an HSP with a molecular mass of 70,000 daltons. In mammalian systems, HSP110, HSP90, HSP70, HSP60, HSP56, HSP47, HSP40, and HSP27 have been identified. The most extensively studied HSPs with respect to their role in thermotolerance and cellular heat sensitivity are HSP27, HSP70, and HSP90. In mammalian cells, good correlations have been reported for thermotolerance development and (1) HSP70 synthesis, (2) HSP27 synthesis, and (3) HSP27 phosphorylation. Enhanced thermosensitivity has been shown to result from reduction of the level of HSP70, whether by competitive inhibition of HSP70 gene expression or by microinjection of cells with antibodies against HSP70. Reduction of the cellular HSP90 levels has also been shown to increase thermosensitivity in mouse L cells, whereas an increased HSP90 expression resulted in stable heat resistance in Chinese hamster ovary (CHO) cells.63 Transfection of cells with genes coding for HSP2764 or HSP70,65,66 resulting in constitutive expression of the genes, confers stable heat resistance. Transfection of rat cells with a gene coding for a homologue of the mammalian HSP60 also confers heat resistance to the transfected cells (Burgman and colleagues, unpublished results). These data provide direct evidence for a causal relation between the expression of HSPs and heat resistance.
Chronic thermotolerance (thermotolerance that develops during long-duration, mild HT at approximately 41° C) may be different from acute thermotolerance, which occurs after heat shock and incubation before heating at high hyperthermic temperatures at or higher than 43° C). Chronic-type thermotolerance is not always observed in all mammalian cell lines. Studies with a range of human cell lines have shown that, unlike rodent cells, the human cells did not develop chronic thermotolerance during mild HT. In HeLa cells, this absence of thermotolerance may be related to cell-cycle progression.67 In other studies, the lack of thermotolerance did not appear to be linked directly to cell-cycle progression, as shown by cell-cycle analysis and the use of plateau-phase cultures.68,69 More research is needed to better understand the development and decay of acute and chronic thermotolerance in human tumors.
Regulation of Heat Shock Response
As described earlier, the heat shock response refers to the increased transcription of a set of genes, the heat shock genes, when cells are subjected to heat stress. It is a highly conserved process that occurs in all organisms from bacteria to human. In eukaryotes, the response is mediated by a sequence-specific DNA binding protein termed HSF1.70 In its trimeric form, HSF1 binds tightly to multiple copies of a highly conserved sequence motif (nGAAn) in the promoter region of heat shock genes; these HSF-binding sites are termed the heat shock elements (HSEs).71,72 In unstressed mammalian cells, HSF1 is maintained in a monomeric, non-DNA binding form. Upon heat shock, HSF1 assembles into trimers, binds to HSEs with high affinity, becomes hyperphosphorylated, and activates the transcription of the heat shock genes. Transcriptional activation of these genes, in turn, increases the synthesis of HSPs. During post–heat-shock recovery, the HSF1 trimers dissociate from the DNA, and are eventually converted to the inactive, non-DNA binding monomers. Several other models, similar to the previously described model, have also been proposed.18,73
Although the importance of HSF1 in the regulation of mammalian heat shock genes is well-established,70 data from multiple laboratories obtained using various rodent and human cell lines indicate that the activation of HSF1 by itself is not sufficient for the induction of HSP70 messenger RNA (mRNA) synthesis,62,74–80 and that other factors may be involved in the regulation of HSP70 transcription. Subsequent studies of Rat-1, hamster HA-1, and HeLa cells provided evidence of the plausible involvement of a negative regulator,62,81 initially termed constitutive HSE binding factor (CHBF). Upon heat shock, the cellular level of the HSE binding activity of HSF1 rapidly increases (“activation of HSF1”) and that of CHBF rapidly decreases (“inactivation of CHBF”). When HSP70 transcription returns to its pre–heat-shock level during post–heat-shock recovery, the disappearance of the HSF1-HSE binding activity again parallels the reappearance of the CHBF-HSE binding activity.62,82 Interestingly, in rat, mouse, and human cells treated with sodium salicylate or sodium arsenite, which elicits considerable HSF1-HSE binding activity but shows no reduction in CHBF-HSE binding activity, the induction of HSP70 mRNA synthesis was not observed.62,77,80,83 Thus, data from studies of a wide variety of rodent and human cells all support the notion of a negative regulatory role of CHBF in HSP70 induction.
Subsequently, CHBF was purified and identified as the Ku protein, a heterodimer of Ku70 and Ku80,81 allowing more direct testing of the hypothesis that CHBF/Ku acts as a negative regulator in the heat induction of HSP70. To carry out such experiments, Li and colleagues established Rat-1 cells that stably and constitutively express human Ku70 and/or Ku80, and found that overexpression of the human Ku70 by itself, or Ku70 and Ku80 jointly, specifically inhibits heat-induced HSP70 expression without affecting induction of other HSPs or activation of HSF1.84,85 How does Ku70 specifically suppress HSP70 expression? There are at least four non–mutually exclusive possibilities: (1) Ku70 may bind directly to sites located in the HSP70 promoter and repress transcription; (2) Ku may regulate HSF1 binding to the HSP70 promoter; (3) Ku may affect the interaction among other transcription factors and their binding to the HSP70 promoter; (4) overexpression of Ku70 could result in altered chromatin structure, inhibiting the access of positive factors to their respective regulatory elements in the HSP70 promoter. Related to the second possibility is the recent finding that in vitro translated Ku70/Ku can bind to HSF1 and displace it from HSE.86,87 Thus the existing evidence strongly supports a role of Ku in HSP70 gene regulation.
Interaction of Heat With Radiation
Heat interacts with radiation in a more than additive way. This synergistic interaction of heat and radiation is interpreted as a heat-induced sensitization of cells to radiation, termed heat radiosensitization, or thermal radiosensitization. Heat radiosensitization can be quantified using a thermal enhancement ratio (TER), defined as the ratio of radiation doses with and without heat to produce the same biologic effect (isoeffect TER). In some cases, TERs are expressed as a ratio of the biologic effects for the same radiation dose, with and without heat (isodose TER), or as a ratio of the D0’s of the radiation survival curves. Usually, TER increases with increasing heat dose. The maximal interaction is observed when heat and radiation are given simultaneously. The TER decreases with an increasing time interval between heat and radiation (Fig. 74-4). When radiation precedes HT, sensitization is no longer detectable 2 to 3 hours after radiation. When HT precedes radiation, cells can be sensitized for up to several hours.
The therapeutic gain factor (TGF) is defined as the ratio of the TER in the tumor to the TER in surrounding normal tissue. Maximal heat radiosensitization is achieved when heat and radiation are applied simultaneously, but a therapeutic gain (more damage to tumor than to normal tissue) may not always be obtained under these conditions, especially if tumor and normal tissue are heated to the same degree.88,89 In some cases, the TGF may increase when time intervals are introduced between radiation and heating.
The combination of heat and radiation does not result in an enhancement of heat lesions; thus, no radiation-induced heat sensitization takes place and the synergistic effect is most likely due to radiosensitization by heat.56 The primary target for radiation is believed to be DNA, whereas protein denaturation and aggregation seem to be involved in thermal cell killing.42,90 DNA damage (e.g., breaks) can be detected at low lethal doses of x-rays (≤1 Gy), whereas no DNA breaks are found at low lethal heat doses.91 The combination of radiation and HT generally does not result in more initial DNA breaks than are observed for radiation alone.90,91 Instead, HT appears to inhibit the rejoining of radiation-induced DNA strand breaks91–93 and the excision of damaged bases94 in a heat-dose–dependent manner.91,93 This effect may be caused by (1) heat-induced inactivation of DNA repair enzymes88,95; or (2) alteration of the chromatin structure resulting from protein denaturation and aggregation, which causes decreased accessibility of the damaged sites to the repair machinery.44,92,94,96 Using cell lines in tissue culture, researchers found a good correlation between the amount of heat-induced nuclear protein aggregates and the extent of thermal radiosensitization.95,97
The effect of thermotolerance on heat radiosensitization has also been a topic of extensive investigation. Studies using tissue-cultured cell lines have shown that thermoradiosensitization could be affected by thermotolerance development.98,99 Because it has been reported that HSPs might be involved in the protection against nuclear protein aggregation as well as in facilitated disaggregation,46,48 the presence of these proteins might indirectly be responsible for the thermotolerance effect on protein aggregation and as such affect DNA repair and cellular radiosensitivity. Rodent cells transfected with human HSP70 showed protection against heat-induced nuclear protein aggregation that was reflected in the extent of thermal radiosensitization.95 Transfection with HSP27 resulted in an accelerated nuclear protein disaggregation and was paralleled by an accelerated decline of thermal radiosensitization. These observations point to the importance of HSPs as a regulator in protein denaturation, aggregation, and disaggregation, thereby modulating the heat effects on DNA repair. As such, the expression of HSPs may explain the effect of thermotolerance on the extent and disappearance of thermal radiosensitization and could have a major effect on the choice of sequence and spacing of heat and radiation in clinical practice. Elevated HSP expression should be either avoided, if occurring in the tumor, or exploited, if occurring in the normal tissue at risk during treatment.
Chronic thermotolerance has been found to affect heat radiosensitization in some cell lines,17,93,99,100 although for other cell lines no effect of thermotolerance was observed at the level of heat radiosensitization.99,101 For example, chronic thermotolerance development in CHO cells inhibited further sensitization to radiation.100 This lack of further radiosensitization was also found in HeLa cells when no chronic thermotolerance developed.67 Interestingly, long-duration, mild HT (as low as 41° C) given concurrently with low-dose-rate irradiation can cause extensive radiosensitization and eliminate the low-dose-rate irradiation sparing effect.69,88,102,103 This thermoradiosensitization to low-dose-rate irradiation occurred for protracted heating times and did not appear to be inhibited for prolonged heating times, supporting the absence of a thermotolerance process in radiosensitization.69,102 The mechanism underlying this low-temperature radiosensitization may very well be different from the one discussed in the previous section on the mechanism of heat radiosensitization because DNA repair rates are often found to be higher at 41° C104–106 and heat-induced protein aggregation in the nucleus is only limited (Stege and associates, unpublished results). Effects of heat on cell-cycle progression or changes in fidelity of repair may be possible explanations of radiosensitization by long-duration, low-temperature HT.
Heat and Chemotherapy
Although the combination of HT with radiation has been the focus of more attention, there is an equally strong rationale for combining HT with chemotherapy. Moderate or even mild HT (such as 39° C) enhances cell killing in vivo of a number of chemotherapeutic agents, such as cyclophosphamide, melphalan, mitomycin C, cisplatin, doxorubicin (DOX), bleomycin, and the nitrosoureas. A variety of mechanisms may account for increased chemotherapeutic effect at elevated temperatures, including altered pharmacokinetics or pharmacodynamics, increased DNA damage, decreased DNA repair, reduced oxygen radical detoxification, and increased membrane damage.107 Concentrations of agents that are not normally toxic at normal body temperature can become cytotoxic at 39° C, and, in some cases, HT may partially overcome some types of drug resistance.
Heat and Liposomes
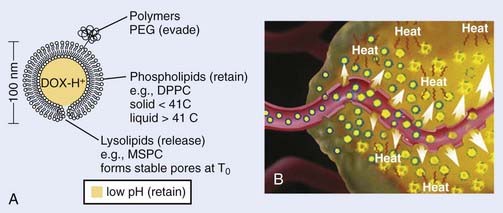
It is well recognized that when systemic chemotherapy is used for the treatment of solid tumors, significant normal tissue toxicities commonly occur when therapeutic levels of drug at the tumor site are achieved.108 Liposomes have been identified as one of the promising carriers for therapeutic agents to improve drug delivery in the treatment of cancer.109,110 Conventional liposomes have been clinically evaluated and approved in a variety of diseases111–113 and HT has been shown to increase extravasation of conventional liposomes within tumor tissue.114 Early in 1978, Yatvin and colleagues proposed the use of HT with temperature-sensitive liposomes to allow increased control over the release of encapsulated drugs at the diseased site,115 and their efforts have stimulated many additional studies.116 In 2000, Dewhirst and collaborators developed a novel thermosensitive, DOX-containing lipid formulation optimized for rapid drug release under mild hyperthermic temperatures (39° C to 40° C) readily achievable in the clinic (Fig. 74-5).113,117,118 Their in vitro studies clearly demonstrate advantages that the new lipid composition has compared with existing liposome formulation; the characteristics of attainable and narrow triggering temperature, rapid release of drug, and high accumulative drug release appear to offer potential advantages that could lead to an increased therapeutic effect over traditional chemotherapy and current liposomal and other drug carrier and delivery systems. Furthermore, in vivo studies demonstrated that this novel liposome, in combination with mild HT, was significantly more effective than free-drug or current liposome formulations at reducing tumor growth in a human squamous cell carcinoma xenograft line (FaDu), producing 100% complete regressions in 11 tumors lasting up to 60 days post-treatment.113,117 To monitor the liposome concentration distribution and drug release in vivo, magnetic resonance imaging (MRI) contrast agent, MnSO(4) and DOX were encapsulated together within this thermosensitive liposomes.119 The MRI clearly showed that the thermally sensitive liposome was only accumulated at the periphery of the tumor, concordant with the release temperature of this formulation (39° C–40° C). Because the liposomal drugs accumulate only in perivascular regions in tumors after intravenous injection, tumor cells in deeper tissue layers cannot be killed. To circumvent this deficiency, Dewhirst and collaborators decided to target tumor microvessels and therefore stop nutrient supply to deeper tumors by constructing a DOX–encapsulated, lysolecithin-containing thermosensitive liposome (LTSL). After DOX-LTSLs in combination with HT treatment, the blood flow of FaDu tumor implanted in dorsal skinfold chamber was significantly decreased, whereas only minor reductions in normal microcirculation in subcutaneous tissues were observed.120 Furthermore, a real-time evaluation of therapeutic protocols in association with outcome on an individual subject basis could be achieved by noninvasively monitoring the temporal and spatial patterns of DOX/MnSO(4)-LTSLs delivery with MRI.121
Heat Shock Proteins and Cancer Immunotherapy
Although HSPs have been intensively studied at the level of transcription and translation for mammalian cells for more than 2 decades, a novel and physiologically important role for these highly conserved gene products has only become evident in the past several years. In the early 1990s, Srivastava and colleagues found that a tumor rejection antigen, isolated by biochemical fractionation of tumor cells, was an HSP (grp94/gp96).122–125 Since then, convincing evidence has accumulated that the HSPs derived from a given cancer, including HSP70, HSP90, and grp94/gp96, can elicit protective immunity specific to that particular cancer. HSPs derived from normal tissues do not have such effect against any cancer tested. In recent years, the immunogenicity of HSP preparations from tumors has been seen in different experimental tumor systems of distinct histologic origins, ranging from chemical- or ultraviolet (UV)-induced tumors to spontaneous tumors.126–128 Immunization of mice and rats with HSP-peptide complexes leads to protective and specific immunity against cancer from which the HSP-peptide complexes are derived.124 This phenomenon has been shown in mice124 and rats129 in a wide array of tumor types, including fibrosarcoma,122 hepatocarcinoma,129 lung carcinoma, melanoma, colon carcinoma,127 squamous carcinoma,130 leukemia,131 and in prophylaxis,122,129 as well as in therapy of pre-existing diseases.127 Two high-molecular weight HSPs, HSP110 and grp170 derived from CT26 and Meth A tumors, were found to induce an antitumor response against these tumors.132–134 Furthermore, it has been demonstrated that the immunization of mice resulted in the induction of memory T cells.130 Taken together, these data suggest that HSPs prepared from tumors can serve as tumor vaccines, and that HSPs may play a central role in the host’s immune system. Currently, one HSP, the endoplasmic reticulum stress-response protein gp96, is undergoing clinical trials for cancer treatment and has yielded promising results, including the induction of antitumor immunity and some benefit for patients when administered as part of a multidose regimen.135 A recent phase III comparison of Vitespen, an autologous tumor-derived HSP gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma, shows that patients with less advanced disease (M1a and M1b) benefit from increasing doses of vaccine treatments.136 Future advances in HSP-based immunotherapy will be aided by an understanding of the mechanisms by which HSP-peptide complexes induce innate and adaptive immunity to tumor cells and target the killing of primary and metastatic cancer cells.137
Hyperthermia and Gene Therapy
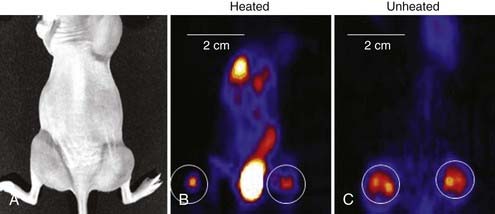
Another new application of HT is its use in gene therapy to provide both spatial and temporal control over gene expression governed by heat shock or other stress-inducible promoters.138–141 As one example, Huang and colleagues transfected cells with adenovirus vectors containing an HSP70 promoter and genes for green fluorescence, interleukin 12, or tumor necrosis factor alpha.142 Experiments with heat-induced green fluorescence protein showed detectable expression at 3 hours, reaching a peak at 18 to 24 hours and disappearing by 72 hours after heating. A modest heat shock of 42° C for 30 minutes increased interleukin-12 gene expression by more than 13,000-fold over baseline and increased tumor necrosis alpha gene expression by 6.8 × 105-fold.142 To optimize the application of heat-activated gene-RT in the clinic, techniques are being investigated to noninvasively monitor the transgene activation. Li and colleagues first used micro–positron emission tomography (microPET) imaging to monitor the improvement in viral vector distribution after a mild heat shock 6 hours after administration of virus (Fig. 74-6).143 Rogers and colleagues further used microPET imaging to noninvasively monitor the heat-inducible suicide gene expression in mice bearing head and neck squamous cell carcinoma xenografts.144 Recently, Chrit and colleagues investigated in vivo spatiotemporal control of transgene activation under the regulation of a heat-inducible promoter using magnetic resonance temperature imaging for image-guided, high-intensity focused ultrasound (US).145
Hyperthermia and Tumor Hypoxia
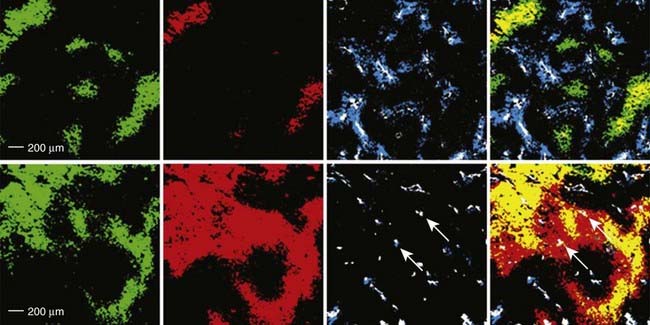
Tumor hypoxia is not only one of the major factors contributing to RT resistance of solid tumors that leads to adverse treatment outcome, but also is related to disease progression and metastasis.146–149 Mild HT (<42° C) is believed to dilate tumor blood vessels and increase tumor oxygenation, and so enhance tumor radiosensitivity.150 Clinical and experimental studies showed that mild HT could increase the overall tumor pO2 level in rodent,151 canine,152 and human tumors,153,154 and the reoxygenation can even last for 1 to 2 days after heating in some tumor types.150 Recently, the change in tumor hypoxia induced by mild HT was investigated at the microscopic level by dual hypoxia marker immunohistochemistry technique in a human colorectal adenocarcinoma xenograft model (HT29).155 It was found that although the overall hypoxic fraction was significantly decreased during heating, mild HT induced both increases and decreases in tumor hypoxia in different regions of the tumor (Fig. 74-7).155 Specifically, mild HT decreased hypoxia in the regions with relatively well-perfused blood vessels, but increased hypoxia in regions that were poorly perfused. These data implied that the changes in tumor oxygenation depend on the vasodilation ability of tumor microvessels in response to HT.
Physical Principles
Mechanisms of Heating
Thermal Conduction
In the simplest form of HT, tissue may be heated by circulating externally preheated blood through the tissue, by placing a heated surface (e.g., water bolus pad) on the skin or in natural body cavities, or by interstitially implanting hot sources in wires, needles, or catheters into the target tissue. It is generally not possible to heat well-perfused, normal tissue more than 3 to 5 millimeters from a hot surface.156
A good approximation for describing heat transfer in biologic tissue is given by the bioheat transfer equation (BHTE), derived in 1948 by Penne157:
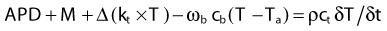
in which Δ is the vector gradient operator; tissue thermal conductivity (kt) quantifies the tissue’s ability to conduct heat; ωb and cb quantify the perfusion rate and specific heat capacity of blood; ct and ρ are the specific heat and mass density of tissue; APD represents the absorbed power density (APD) or external heat input to the tissue; M is the heat generated from tissue metabolism (generally negligible compared with other heat inputs); and T and Ta are the tissue and arterial blood temperatures, respectively, at time t. The first two terms represent heat inputs to the tissue volume; the third and fourth terms describe heat transfer kinetics caused by thermal conduction and convection (blood flow), respectively; and the right side of the equation specifies the net heating effect in terms of the rate of change of temperature with time (δT/δt). Alternative theoretical models have been studied to describe the heat transfer characteristics of tumors more accurately by accounting for “thermally significant” blood vessels,156,158 but the BHTE serves as a good starting point. Review articles are available on thermal modeling and HT treatment-planning issues.159–161
Electromagnetic Power Deposition
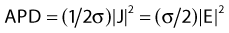
in watts per cubic meter, in which J = σE is the induced current density. This APD is closely related to the more commonly used power deposition rate or specific absorption rate (SAR) by the formula:
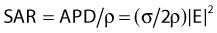
in which ρ is the mass density of tissue in kilograms per cubic meter.
For both RF and MW radiation, APD and SAR decrease with depth in tissue. The APD for the simplest case of plane-wave radiation is plotted as a function of penetration depth into muscle tissue in Fig. 74-8. At higher frequencies, power is deposited more superficially, whereas lower frequencies provide deeper penetration. However, for practical HT applicators, the tissue penetration may be much less than this because of coupling problems at the applicator-tissue interface, tissue heterogeneities, and nonuniform beam profile in the near-field region of HT applicators that are small compared with the wavelength.
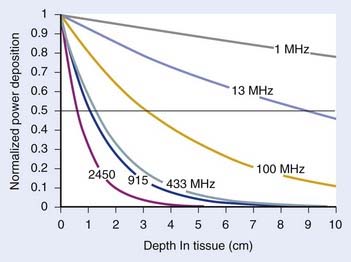
For frequencies used in HT, the wavelength (λ) of EM energy in soft tissue varies from approximately 3.4 cm at 10 MHz to less than 4 cm at 1000 MHz. The minimum spot size for focusing power deposition is approximately λ/2, or approximately 20 cm at the lowest RF frequencies typically used (100 MHz) and approximately 2 cm at the higher MW frequencies. Thus, lower RF frequencies penetrate well but affect large regions of the body, whereas higher MW frequencies may be localized effectively in tumor-sized tissue volumes. Compilations of electrical conductivity, dielectric constants, wavelengths, and penetration depths of EM waves in air, muscle, and fat are readily available.161,162
Ultrasound Power Deposition
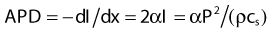
in watts per cubic meter, and again SAR is APD/ρ in watts per kilogram.
Fig. 74-9 shows the penetration depth characteristics of US in homogenous muscle tissue as a function of frequency, assuming well-behaved plane wave radiation. Higher frequencies result in more superficial localization of power and lower frequencies result in deeper penetration. For practical HT transducers, the target volume usually appears in the transducer near field, where there are marked fluctuations in beam intensity both longitudinally in the direction of propagation as well as across the wavefront, as opposed to much smoother inverse-square-law longitudinal decay of intensity in the far field. Thus, reflections, scattering, and thermal conduction within the tissue are relied on to smooth the temperature distribution that results from the peaky SAR. Nonfocused applicators operating at approximately 3.5 MHz can heat tumors up to 4 to 6 cm in depth.163 Nonfocused applicators operating at lower frequencies generally produce increased pain because of unavoidable absorption in underlying bone. Deeper penetration is made possible by directing the US beams to avoid bone and air interfaces, using a larger acoustic window at the surface, and focusing the US beams to increase the power density at depth relative to that at the skin surface and critical normal tissues outside the focal volume.
The characteristic acoustic impedance Z = ρcs is an important parameter in determining the behavior of US at tissue interfaces. The Z’s of most soft tissues are quite similar, so there is little reflective loss during the propagation of US from one soft tissue to another. However, the Z values for bone, air, and lung are considerably different from those of soft tissues, causing almost complete reflection at soft tissue–gas interfaces and both reflection and rapid absorption of the transmitted portion of the wave at soft tissue–bone interfaces.164 A tabulation of acoustic properties of tissues and more in-depth coverage of US interactions with tissue are available.161,162,165
Heating Equipment and Techniques
Local Heating—External Sources
A large development effort has gone into producing local heating devices with improved control of power deposition pattern. EM techniques are generally used for superficial tumors less than approximately 3 cm in depth, such as chest-wall recurrence of breast carcinoma, whereas US beams are useful for somewhat deeper tumors up to approximately 6 cm deep.161,166–169
Electromagnetic Techniques for Superficial Heating
The simplest, most reliable applicator used in clinical HT has been the single-aperture MW waveguide. A MW radiator mounted within a waveguide (a tubular or rectangular structure for guiding EM waves) radiates EM waves out the waveguide opening (aperture). Commercial applicators have been available in a variety of sizes from 7.5 to 24 cm on a side, normally operated at 915 MHz or approximately 430 MHz as these are the Industrial, Scientific, and Medical band frequencies allowed for use with no power restrictions in the United States, and Europe and Asia respectively. The effective heating area, or the area under the applicator producing 50% or greater of the maximal SAR, typically covers only 30% to 60% of the applicator face, normally with high-SAR “hot spots” located centrally, surrounded by a “cool” periphery.170,171 These single-aperture waveguide devices are useful for only small, superficial tumors up to approximately 3 cm in diameter.8 One group has placed variable absorption bolus pads in front of the aperture to reduce SAR centrally and achieve a larger area of effective heating.172 The HTS-100 Lens Applicator has movable metal plate “vanes” within the concave opening of a single waveguide horn aperture to form convergent 430-MHz beams for heating small tumors up to 3 to 5 cm deep.173
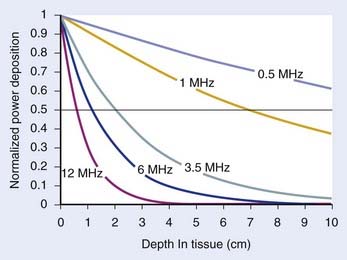
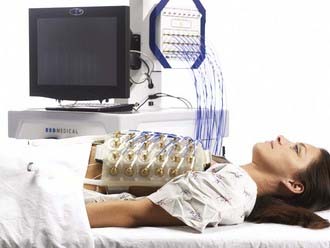
A variety of approaches have been taken to build applicators capable of heating larger areas, such as computer-controlled motorized scanning of one or two spiral microstrip MW antennas repetitively over the target surface174–176 with thermal feedback control systems. Alternatively, several groups have developed multiaperture array waveguide applicators for large-area superficial disease such as the Microtherm 1000 (Labthermics Technologies, Urbana, IL)—a 16-element, 915-MHz waveguide planar array,177 and a somewhat deeper heating array of 433-MHz Lucite Cone Applicators.178,179 Other more flexible tissue-conforming, large-area MW array applicators have been constructed and used clinically, including a 433-MHz contact-flexible microwave applicator180,181; a hinged array of 433-MHz current sheet applicators182; a 915-MHz, 25-element spiral microstrip conformal array183; and subsequent commercial implementations (Fig. 74-10)184; and the largest 915-MHz conformal microwave array (CMA) applicator.168,185 Fig. 74-11 shows a 6-mm thick water bolus vest and flexible printed circuit board CMA applicator being set up on a patient with chest-wall recurrence of breast carcinoma extending across the left upper back and shoulder. The patient is shown at right with an elastic overgarment to secure the applicator in place, allowing the patient freedom to stand, sit, or lie down during heat treatment while providing effective temperature control of the skin surface.186 This applicator is essentially transparent to photon and electron radiation and thus may be used to apply heat simultaneously with radiation for potentially much higher TER.187–189 Recent developments are beginning to produce EM superficial heating applicators with the capability of noninvasive thermal monitoring of surface190 and subsurface191–194 temperature distributions for feedback control of multiple antenna arrays.
Ultrasound Techniques
Early efforts to apply US for superficial heating applications used single-round disk “piston” style transducers operating at 0.5 to 3.5 MHz.195 These devices were capable of heating only small, superficial lesions less than 3 cm in diameter.8 Later, multitransducer arrays were constructed to increase the lateral extent and control of heating. The Sonotherm 1000 (Labthermics Technologies, Urbana, IL) is one such device with 16 individual transducers arranged in a 15-cm square 4 × 4 element planar array coupled to the patient with an attached, degassed water bolus bag.163 This device has been used successfully to heat tumors up to 4 to 6 cm in depth.
Another approach uses arrays of focused transducers. The scanned focused US system uses a small number of low-frequency transducers (≤2 MHz) that produce a small focal spot at depth that is mechanically scanned rapidly around the tumor volume.196 The addition of less-penetrating 4-MHz transducers and a patient pain feedback button to turn off power for short segments of the scan path improved the heating uniformity of this scanning US technique and yielded higher average tumor temperatures.197 Another innovative US system has a computer-controlled scanning reflector for distributing US energy at two different frequencies (e.g., 1 and 5 MHz) over large surface areas.198 With only water and plexiglas over the tumor surface, this system allows simultaneous HT and RT for potentially much higher TER.199–201 Development of high-intensity focused US for heating small- to medium-size regions at depth continues, with most commercial development supporting thermal ablation applications.202,203
Local Heating—Interstitial Sources
For applications requiring precise localization of heat in regions that are too difficult to access externally, there are at least nine distinctly different interstitial heating technologies. Excellent reviews are available.166,204–206
Microwave Antennas
Interstitial MW antennas are usually constructed from flexible or semirigid coaxial cable less than 1.5 mm in diameter that fits snugly inside afterloading plastic implant catheters. The most common types are dipole antennas, which can be used in phased arrays to enlarge the heated volume,207–209 and helical coil antennas, which tend to restrict heating more effectively to the region immediately surrounding the active coil length of the antenna.210,211 Frequencies between 433 and 2450 MHz are normally used for interstitial MW heating. Radial penetration of heating may be extended by several millimeters with air or water cooling of the implanted antenna surface, but generally multiple antennas required for heating larger tumors up to 5 cm across should be placed within 1.5 to 2 cm from each other.212 Implantable MW antennas have been most useful in high-perfusion tissues or when nonparallel or flexible catheter implants are required. Recent developments have produced interstitial MW antennas with integrated MW radiometry for real-time monitoring of tissue temperature around each antenna.213,214
Radiofrequency Electrodes
RF electrode systems heat tissue with resistively or capacitively coupled electric currents between implanted metal electrodes. Initial work used arrays of implant needles up to 20 cm long spaced 1 to 1.25 cm apart connected to a single RF power amplifier operating at less than 1 MHz. Subsequent systems used time-sequenced computer controlled localized current field (LCF) sources that distributed power to multiple electrode pairs around the implant array.204 Systems with segmented RF electrodes,215,216 printed circuit board implant templates to simplify power connections, and computer-control programs allow simultaneous long-duration heat and brachytherapy.204,217 At frequencies above 10 MHz, current may be coupled capacitively through a thin layer of insulating plastic, resulting in a more uniform distribution of heating current, as in the 64-channel, 27.12-MHz capacitively coupled RF (CC-RF) electrode system.216,218,219 For both LCF and CC-RF systems, the radial falloff of power deposition is extremely steep so that implant spacing is limited to less than 1.25 to 1.5 cm for HT applications. Methods of cooling the electrode surface and driving multiple electrode sets have proven useful for extending the effective heating volume, especially for thermal ablation applications in which higher temperatures can be tolerated near the sources.212
Hot Source Techniques
Because the penetration of thermally conducted heat energy is limited, hot source techniques are appropriate only for tissues with low to moderate perfusion that can be implanted with closely spaced arrays of implants—generally no more than 1.0 to 1.25 cm apart. However, heating length is almost unlimited. Hot source techniques include hot water perfusion through implanted catheters,204,217 resistive wire implants heated by computer-controlled direct-current voltage sources,220 and ferromagnetic implants (FMIs) heated by an externally applied magnetic field at less than 1 MHz.221–223 An advantage of the FMI approach is that the implants can be made of special high-permeability alloys that operate near their Curie point transition from magnetic to nonmagnetic such that they automatically thermoregulate over a narrow range of temperature.224,225 The ferromagnetic material providing this controlled temperature can be a needle or rod, 5- to 10-mm long cylindrical seeds interspersed with 5-mm long or less radioactive brachytherapy sources for simultaneous thermobrachytherapy, or ferromagnetic fluids or nanoparticles that are either inserted directly in the tumor in one or more needle tracks or are delivered systemically.226,227 The FMI techniques simplify reheating of permanently implanted seeds,228 rods,229 or nanoparticles230 because no externalized connections are required for either power or thermal monitoring of the thermoregulating implant material.
Ultrasound Radiators
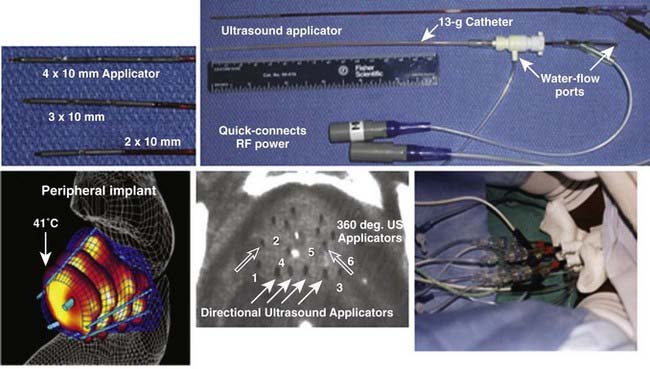
Interstitial US applicators appear close to revolutionizing interstitial HT (IHT) delivery for many sites. Linear arrays of 0.5- to 1-cm long tubular transducers have been constructed either with water cooling231 or direct coupled sources with an open central lumen to accommodate brachytherapy sources for simultaneous thermobrachytherapy.232 Implant spacing up to 2.5 cm apart should be possible for the water-cooled version. These applicators allow precise power control along the implant length as well as radially directional heating capability. Fig. 74-12 shows an example of a clinical treatment of prostate with an array of different-length US array applicators that fit inside 13-g brachytherapy catheters for highly steerable power deposition. Implantable US arrays have been used for treatment with real-time, noninvasive MRI monitoring of both temperature and tissue property changes during heating.233–235