Chapter 25 Hypersensitivity (Type III)
• Immune complexes are formed when antibody meets antigen. They are removed by the mononuclear phagocyte system following complement activation. Persistence of antigen from chronic infection or in autoimmune disease can lead to immune complex disease.
• Immune complexes can trigger a variety of inflammatory processes. Fc–FcR interactions are the key mediators of inflammation. Most importantly, Fc regions within immune deposits within tissues engage Fc receptors on activated neutrophils, lymphocytes, and platelets to induce inflammation. During chronic inflammation B cells and macrophages are the predominant infiltrating cell type, and activation of endogenous cells within the organ participates in fibrosis and disease progression.
• Experimental models demonstrate the main immune complex diseases. Serum sickness can be induced with large injections of foreign antigen. Autoimmunity causes immune complex disease in the NZB/NZW mouse. Injection of antigen into the skin of presensitized animals produces the Arthus reaction.
• Immune complexes are normally removed by the mononuclear phagocyte system. Complement helps to disrupt antigen–antibody bonds and keeps immune complexes soluble. Primate erythrocytes bear a receptor for C3b and are important for transporting complement-containing immune complexes to the spleen for removal. Complement deficiencies lead to the formation of large, relatively insoluble complexes, which deposit in tissues.
• The size of immune complexes affects their deposition. Deposition of circulating, soluble immune complexes is limited by physical factors, such as the size and charge of the complexes. Small, positively charged complexes have the greatest propensity for deposition within vessels. Large immune complexes are rapidly removed in the liver and spleen.
• Immune complex deposition in the tissues results in tissue damage. Immune complexes can form both in the circulation, leading to systemic disease, and at local sites such as the lung. Charged cationic antigens have tissue-binding properties, particularly for the glomerulus, and help to localize complexes to the kidney. Factors that tend to increase blood vessel permeability enhance the deposition of immune complexes in tissues.
Immune complex diseases
Immune complex formation can result from:
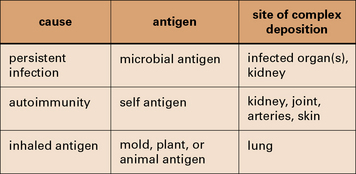
Fig. 25.1 Three categories of immune complex disease
This table indicates the source of the antigen and the organs most frequently affected.
Persistent infection with a weak antibody response can lead to immune complex disease
The combined effects of a low-grade persistent infection and a weak antibody response lead to chronic immune complex formation, and eventual deposition of complexes in the tissues (Fig. 25.2). Diseases with this etiology include:
Immune complexes can be formed with inhaled antigens
Immune complexes may be formed at body surfaces following exposure to extrinsic antigens.
• farmer’s lung, where there are circulating antibodies to actinomycete fungi (found in moldy hay); and
• pigeon fancier’s lung, where there are circulating antibodies to pigeon antigens.
Both diseases are forms of extrinsic allergic alveolitis, and occur only after repeated exposure to the antigen. Note that the antibodies induced by these antigens are primarily IgG, rather than the IgE seen in type I hypersensitivity reactions. When antigen again enters the body by inhalation, local immune complexes are formed in the alveoli leading to inflammation and fibrosis (Fig. 25.3).
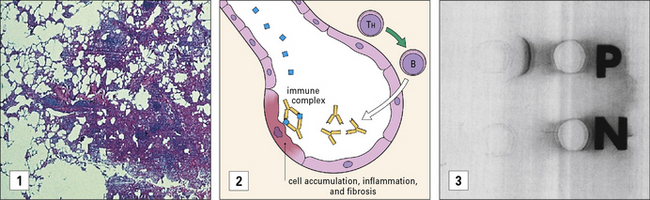
Immune complex disease occurs in autoimmune rheumatic disorders
Immune complex disease is common in autoimmune disease, where the continued production of autoantibody to a self antigen leads to prolonged immune complex formation. As the number of complexes in the blood increases, the systems responsible for the removal of complexes (mononuclear phagocyte, erythrocyte, and complement) become overloaded, and complexes are deposited in the tissues (see Fig. 25.16). Systemic lupus erythematosus (SLE) is the classic disease characterized by immune complex deposition and others include Henoch-Schönlein purpura and primary Sjögren’s syndrome.
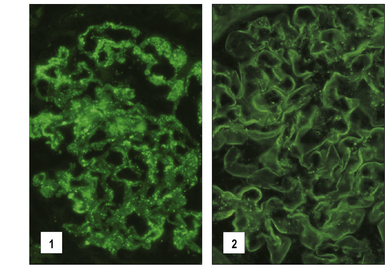
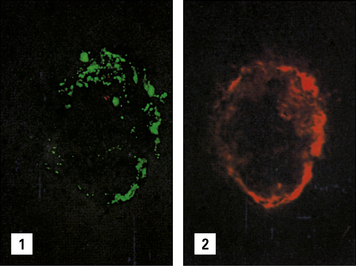
Fig. 25.16 Immunofluorescence study of immune complexes in autoimmune disease
(Courtesy of Dr S Thiru.)
Immune complexes and inflammation
Immune complexes are capable of triggering a wide variety of inflammatory processes:
• they interact directly with basophils and platelets (via Fc receptors) to induce the release of vasoactive amines (Fig. 25.4);
• macrophages are stimulated to release cytokines, particularly tumor necrosis factor-α (TNFα) and interleukin-1 (IL-1), which have important roles in inflammation;
• they interact with the complement system to generate C3a and C5a, which stimulate the release of vasoactive amines (including histamine and 5-hydroxytryptamine) and chemotactic factors from mast cells and basophils; C5a is also chemotactic for basophils, eosinophils, and neutrophils.
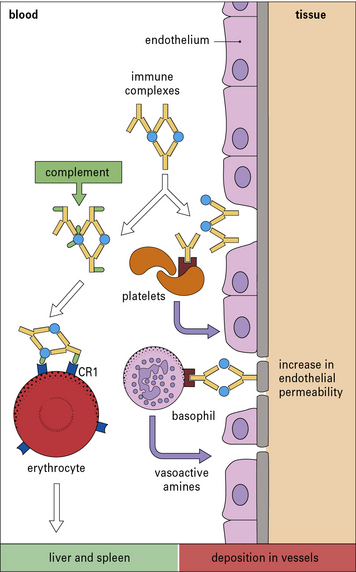
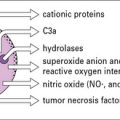
The vasoactive amines released by platelets, basophils, and mast cells cause endothelial cell retraction and thus increase vascular permeability, allowing the deposition of immune complexes on the blood vessel wall (Fig. 25.5). The deposited complexes continue to generate C3a and C5a.
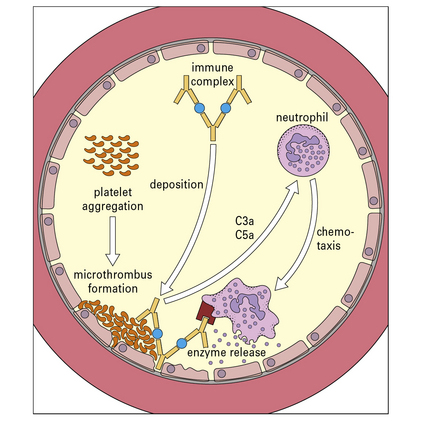
Polymorphs are chemotactically attracted to the site by C5a. They attempt to engulf the deposited immune complexes, but are unable to do so because the complexes are bound to the vessel wall. Therefore they exocytose their lysosomal enzymes onto the site of deposition (see Fig. 25.5). If simply released into the blood or tissue fluids these lysosomal enzymes are unlikely to cause much inflammation, because they are rapidly neutralized by serum enzyme inhibitors. But if the phagocyte applies itself closely to the tissue-trapped complexes through Fc binding, then serum inhibitors are excluded and the enzymes may damage the underlying tissue.
Complement is an important mediator of immune complex disease
• the kidney in various autoimmune glomerular diseases; and
• the skin in autoimmune diseases where cutaneous vasculitis is a feature such as SLE, Sjögren’s syndrome and Henoch–Schönlein purpura.
• the important immune complex solubilizing roles will prevent immune complex deposition until the capacity of the system is exceeded;
• beyond this threshold complexes deposit and activate complement in the tissues, causing pathology.
 be found in SLE.
be found in SLE.Experimental models of immune complex diseases
Experimental models are available for the main types of immune complex disease described above:
• serum sickness, induced by injections of foreign antigen, mimics the effect of a persistent infection;
• the NZB/NZW mouse demonstrates autoimmunity;
• the Arthus reaction is an example of local damage by extrinsic antigen.
Serum sickness can be induced with large injections of foreign antigen
Serum sickness is now commonly studied in rabbits by giving them an intravenous injection of a foreign soluble protein such as bovine serum albumin (BSA). After about 1 week antibodies are formed, which enter the circulation and complex with antigen. Because the reaction occurs in antigen excess, the immune complexes are small (Fig. 25.w1). These small complexes are removed only slowly by the mononuclear phagocyte system and therefore persist in the circulation.
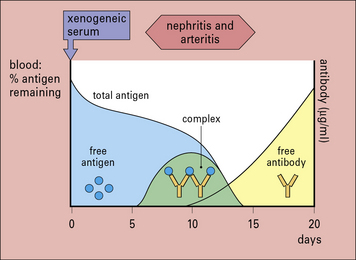
The formation of complexes is followed by an abrupt fall in total hemolytic complement.
Autoimmunity causes immune complex disease in the NZB/NZW mouse
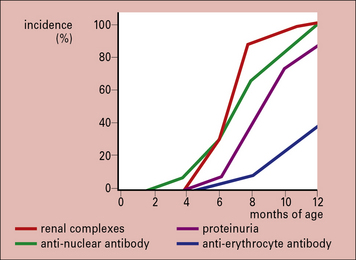
The F1 hybrid NZB/NZW mouse produces a range of autoantibodies (including anti-erythrocyte, anti-nuclear, anti-DNA, and anti-Sm) and suffers from an immune complex disease similar in many ways to SLE in humans. A NZB/NZW mouse is born clinically normal, but within 2–3 months shows sign of hemolytic anemia. Tests for anti-erythrocyte antibody (the Coombs’ test), anti-nuclear antibodies, lupus cells, and circulating immune complexes are all positive, and there are deposits in the glomeruli and choroid plexus of the brain. The disease is much more marked in the females, who die within a few months of developing symptoms (Fig. 25.w2).