Fig. 26.1
Plasma cells obtained at the autopsy of Thomas Alexander McBean, January 1846 (wood engravings made from drawings by Mr. Dalrymple) (Reprinted with permission from Dalrymple J. On the microscopical character of mollities ossium. Dublin Quarterly Journal of Medical Sciences. 1846;2:85–95.)
In light of the autopsy findings, both Dalrymple and Macintyre believed that the disorder that Mr. McBean suffered from was a malignant disease of bone. Bright’s disease was considered in the differential diagnosis because of the albuminous matter in the urine, but there was no dropsy, and the kidneys appeared healthy. The diarrhea, weakness, emaciation, hepatic enlargement, flatulence, dyspepsia, edema of the ankles, puffiness of the face, and large amounts of Bence Jones proteinuria suggest to contemporary readers the possibility of amyloidosis in addition to myeloma. However, autopsy findings of a normal heart and kidneys and “voluminous liver of healthy structure” make the presence of amyloidosis unlikely. Because the lardaceous or waxy changes of amyloidosis in the liver were commonly recognized during the 1840s (although not yet understood), it is unlikely that the gross changes of amyloidosis would have been overlooked if present in Mr. McBean [8].
In 1967, Bristol experimental pathologist John Clamp located Mr. McBean’s death certificate in the General Register Office in London, using dates and descriptions of the patient from the case reports of Drs. Macintyre and Bence Jones. Dr. Clamp suggested that the name multiple myeloma might instead have been McBean’s disease with Macintyre’s proteinuria [3]. However, although Dr. Macintyre first noticed the peculiar heat properties of Mr. McBean’s urine, it was Bence Jones who emphasized its place in the diagnosis of myeloma—“I need hardly remark on the importance of seeking for this oxide of albumen in other cases of mollities ossium”—and who can be credited with developing the first biochemical test for detection of cancer [1, 9]. The modest Dr. Macintyre stated that his “share in this part of the inquiry, it must have been seen, was very humble… I shall be content if I have succeeded in pointing out to future observers, gifted with the requisite qualifications for conducting researches of a higher order, certain definite and distinctive characters by which a peculiar and hitherto unrecorded pathological condition of the urine may be recognized and identified” [4].
Earlier Cases of Multiple Myeloma
Although the first clear description of multiple myeloma did not occur until the 1846–1850 chemical, clinical, and pathological reports of Bence Jones, Macintyre, and Dalrymple described previously (all single-authored publications), the disease has undoubtedly existed for centuries. It seems likely that some of the cases of “mollities ossium” reported in the eighteenth and early nineteenth centuries represent patients who had myeloma. However, without detailed microscopic description of plasma cells such as that provided by Mr. Dalrymple in Mr. McBean’s case, or recognition of a unique disease-associated protein such as that first detected by Drs. Bence Jones and Macintyre, it is not possible to be certain.
When examining old skeletons, sharply demarcated spheroid skeletal lesions that are “purely lytic”—i.e., lacking gross evidence of sclerosis or formation of new bone—are suggestive of multiple myeloma, especially when such lesions are multiple and occur in the proximal long bones and axial skeleton [10]. Two male skeletons with this bony lesion pattern, with estimated ages at death of between 40 and 60 years and dating from 3200 to 500 bc, were identified from among 905 individuals excavated at Thebes-West and Abydos in Upper Egypt [11], while two similarly affected skeletons were found among 2,547 individuals entombed in a rural South German ossuary between 1400 and 1800 ad [12].
Paleopathologists have identified additional ancient bones with features suggestive of multiple myeloma, such as the skeleton of a middle-aged Icelandic female from the eleventh to fifteenth century ad [13], two calvaria from medieval Britain [14], four American Indian skeletons from 200 to 1300 ad [15], and 14 pre-Columbian American skeletons dating back to 3300 bc [16]. The Hunterian Museum of the Royal College of Surgeons in London has in its collection the bones of an approximately 45-year-old Roman soldier with myeloma-like lesions. Suspicious rounded lytic lesions can also be seen in the remains of George Grenville (1712–1770), the Whig Prime Minister whose administration passed the notorious Stamp Act of 1765 that first alienated American colonists from England, and who was autopsied by pioneering Scottish surgeon John Hunter in 1770 [17].
Multiple myeloma with Bence Jones proteinuria occurs spontaneously in contemporary animals [18], raising the possibility that myelomatous lesions might be identifiable in prehistoric nonhuman fossils. Indeed, paleontologists have observed multiple lytic defects without evidence of bony remodeling in a few dinosaur skeletons from the Jurassic and Cretaceous periods, and these have been interpreted by some observers as evidence of an origin of multiple myeloma in the Mesozoic era or earlier [19]. However, caution is indicated in interpreting such ancient specimens.
In any case, it is almost certain that 39-year-old Sarah Newbury, a patient described by distinguished London surgeon Samuel Solly in 1844, had multiple myeloma [20]. Mrs. Newbury had experienced increasing fatigue and, 4 years before her death, was suddenly seized with a violent pain in her back when stooping. “Rheumatic pains” in her limbs occurred a year later, and these progressed to the point where her gait became unsteady. Her limb pain increased greatly after a fall in February 1842, which resulted in inability to lift her right leg, and she was confined to her room. Two months later, her femurs fractured “into a thousand pieces” when her husband, a policeman, lifted her from the fireside to carry her to her bed. Pathological fractures of the clavicles, right humerus, and right radius and ulna followed.
Mrs. Newbury first saw Mr. Solly in consultation in October 1843, but thereafter wrote him a letter asking him not to visit her again because she was vexed that he had prescribed a bitter infusion to try to improve her poor appetite. She was then lost to medical follow-up until she was admitted at the insistence of her husband to St. Thomas’s Hospital in Southwark (South London) on April 15, 1844, in an extremely debilitated state. Over the next few days she was treated with an infusion of orange peel and a rhubarb pill, and an opiate at night if required. Examination of the copious amount of urine she produced revealed a “large quantity of phosphate of lime,” but blood tests could not be performed because Mrs. Newbury was “too suspicious and irritable” to allow them [20]. She died suddenly on April 20, 1844, of “asphyxia” [20].
At autopsy, Mrs. Newbury’s thoracic cavity was reduced to just 4 in. in transverse diameter. The right lung was compressed to about one-fourth of its natural size, and the left lung was decreased to one-half the extent of the right lung because of skeletal changes. The cancellous portion of the sternum (Fig. 26.2) had been replaced by a red substance similar to that seen in Mr. McBean [4]. A peculiar red material had also replaced much of the femur; it ranged in color “from a deep Modena red to a bright scarlet crimson” (Fig. 26.3). Solly examined this red matter with Mr. John Birkett of Guy’s Hospital, a surgeon and anatomist, who described the cells it consisted of as “very clear, their edge being remarkably distinct, and the clear oval outline enclosing one bright central nucleus, rarely two, never more” [italics Solly’s] [20]. John Dalrymple later noted that the microscopic appearance reported by Mr. Birkett “accords very nearly” with his own description of Mr. McBean’s marrow [7]. Solly postulated that the process affecting Mrs. Newbury was inflammatory and had begun with an abnormality of the blood vessels, in which the “earthy matter of the bone is absorbed and thrown out by the kidneys in the urine” [20].
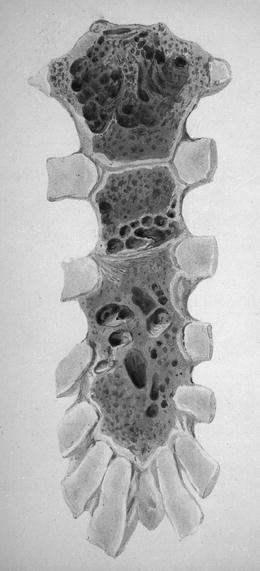
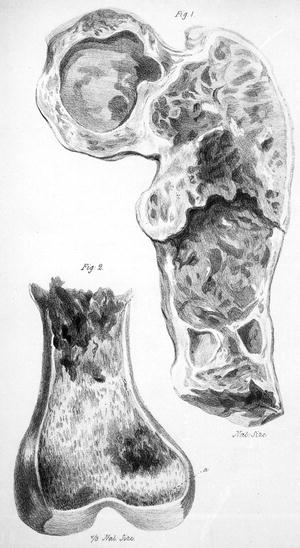
Fig. 26.2
Sternum of Sarah Newbury showing destruction of bone; drawn after her autopsy in April 1844 (Reprinted with permission from Solly S. Remarks on the pathology of mollities ossium with cases. Medico-Chirurgical Transactions of London. 1844;27:435–461.)
Fig. 26.3
Femur of Sarah Newbury showing destruction by myeloma tumor (Reprinted with permission from Solly S. Remarks on the pathology of mollities ossium with cases. Medico-Chirurgical Transactions of London. 1844;27:435–461.)
Other Contributions to Bence Jones Proteinuria and Monoclonal Protein Detection
Although much has been written about the life and scientific work of Henry Bence Jones [2, 21–23], a number of other persons played a part in the evolving story of Bence Jones proteinuria.
In 1846, Johann Florian Heller, an Austrian chemist and physician working in Vienna, described a protein in the urine of a patient that precipitated when warmed a little above 50 °C and then disappeared upon further heating [24]. Although Heller did not recognize the re-precipitation of the protein when the urine was cooled, it seems likely that he was observing a Bence Jones protein. Heller also distinguished this new protein from albumin and casein [24]. R. Fleischer in Germany was the first to use the term “Bence Jones protein” in writing, in an 1880 paper describing a substance with chemical characteristics of Bence Jones protein that he isolated from normal bone marrow [25].
In 1883, Wilhelm Friedrich Kühne, a prominent Berlin physiologist who is best known for coining the term “enzyme,” described finding Bence Jones protein in the urine of a 40-year-old patient from Amsterdam who had died in 1869 after an illness characterized by bone pain, spinal curvature, and cranial neuropathy, possibly from an extramedullary plasmacytoma [26]. Autopsy results were not reported. Kühne isolated the protein and found that the carbon, hydrogen, and nitrogen levels were similar to those described by Bence Jones, and attributed any differences in results between his report and that of Bence Jones to the fact that his preparation was more pure than Bence Jones’s preparation. He named the peculiar protein “albumosurie.”
In 1898, T.R. Bradshaw of Liverpool observed that meals had little or no influence on the amount of Bence Jones proteinuria [27]. There was no nocturnal variation, and Dr. Bradshaw believed that the rate of protein excretion was “pretty constant throughout the 24 h.”
In 1899, A. Ellinger in Germany suggested that there might be an abnormal protein in the blood in patients with myeloma that was similar to the Bence Jones protein, but he was unable to prove this assertion [28].
Waltman Walters at Mayo Clinic in Minnesota described three carefully-evaluated patients with multiple myeloma in 1921, and reported that the quantity of Bence Jones proteinuria in these patients was independent of their oral protein intake [29]. Like Dr. Bradshaw, Walters also noted no diurnal variation. In one patient, intravenous injection of Bence Jones protein appeared to increase the amount of Bence Jones proteinuria. Walters also found Bence Jones protein in the blood of one patient and in the bronchial secretions of another. He concluded that Bence Jones proteins were of endogenous origin, and hypothesized that they were derived from blood proteins through the action of abnormal cells in the bone marrow [29].
The following year—1922—Stanhope Bayne-Jones and D.W. Wilson from Johns Hopkins made 12 preparations of Bence Jones proteins from five patients, two of whom had been included in Dr. Walters’ report [30]. Drs. Bayne-Jones and Wilson immunized rabbits by intravenous injection of the Bence Jones protein, and performed precipitin tests with the Bence Jones protein preparations, concluding that Bence Jones proteins consisted of two groups of similar but not identical proteins.
Beginning in the 1930s, there was considerable debate between proponents of two schools of thought on the origin of Bence Jones protein. The first theory, championed by Adolf Magnus-Levy in Berlin, held that proteinaceous materials found in the urine were the result of overproduction of normal serum proteins by the bone marrow [31]. An alternative view was held by Maxwell Wintrobe and M.V. Buell at Johns Hopkins in Baltimore. In a description of the phenomenon of cryoprecipitation in 1933, Wintrobe and Buell argued that pathological proteins in plasma cell disorders were likely to be distinct from all normal serum components [32].
In the early 1950s, protein chemist Frank W. Putnam at the University of Chicago performed a series of experiments in myeloma patients using 13 C radioisotopes, which helped clarify the origin of Bence Jones proteins. Putnam first showed that the Bence Jones proteins from 18 different patients with myeloma were all unique, though as had been suggested by Bayne-Jones and Wilson, they clustered into two antigenic groups. In 1955, Putnam and his co-worker Sarah Hardy showed that Bence Jones proteins derived directly from the body’s metabolic pool of nitrogen, rather than being a breakdown product of some sort of plasma precursor [33].
In 1956, Leonhard Korngold and Rose Lipari at New York’s Sloan Kettering Institute for Cancer Research and Cornell Medical College Department of Biochemistry formally demonstrated a relationship between Bence Jones protein and the serum proteins of multiple myeloma [34]. As a tribute to Korngold and Lipari, the two major classes of Bence Jones proteins are designated κ and λ.
One hundred and seventeen years after the initial description of the unique heat properties of Bence Jones protein—in 1962—Gerald Edelman and Joseph Gally at the Rockefeller Institute for Medical Research in New York demonstrated that the light chains prepared from a serum immunoglobulin G (IgG) myeloma protein and the Bence Jones protein from the same patient’s urine were identical in all respects: the same amino acid sequence, similar spectrofluorometric behavior, the same molecular weight, identical appearance on chromatography with carboxymethylcellulose and on starch gel electrophoresis after reduction and alkylation, and the same ultracentrifugal pattern—as well as the same thermal solubility [35]. The light chains precipitated when heated to between 40 °C and 60 °C, dissolved on boiling, and re-precipitated when cooled to between 40 °C and 60 °C.
Shortly after Edelman and Gally’s discovery, Norbert Hilschmann and Lyman Craig of the Rockefeller Institute [36] and Koiti Titani and colleagues in Putnam’s laboratory [37] provided the first antibody amino acid sequences, and showed that Bence Jones proteins were not only related to the light chains of gamma globulin but that each light chain was divided into a “variable” or V region, and a “constant” or C region. This structure accounts for the heterogeneity of normal gamma globulins and for antibody specificity and diversity.
Diagnostic Tests for Myeloma: Beyond Bence Jones Proteinuria
Arne Tiselius in Uppsala, Sweden, a 1948 Nobel Laureate in Chemistry, reported an improved method of serum electrophoresis in 1937, which allowed separation of serum globulins into three components: alpha, beta, and gamma [38]. In 1939, Tiselius isolated antibody activity to the gamma fraction [39], while Lewis Longsworth and colleagues at the Rockefeller Institute first noted the classic myeloma “M-spike” in that same year, using Tiselius’ electrophoretic techniques [40].
In 1953, Pierre Grabar and Curtis A. Williams at the Institut Pasteur in Paris described immunoelectrophoresis, which has facilitated the diagnosis of multiple myeloma [41]. Immunofixation or “direct immunoelectrophoresis” was reported by Armine T. Wilson from the Alfred I. du Pont Institute in Wilmington, Delaware, in 1964 [42]. Wilson applied antisera on the surface of the agar immediately after completion of electrophoresis. Immunofixation has proven useful when the results of immunoelectrophoresis are equivocal [43], and is also helpful in the recognition of small monoclonal light chains not detectable by immunoelectrophoresis [44].
Oligosecretory multiple myeloma is defined by the absence of detectable monoclonal proteins in serum and urine using immunoelectrophoresis and immunofixation; it accounts for <5 % of patients with multiple myeloma. In 2001, Arthur J. Bradwell from Birmingham, England, and his colleagues reported an immunological method for detecting imbalance in the concentration of κ and λ serum-free light chains, using specific antibodies that bind only to free light chains, not to light chains bound to immunoglobulin heavy chains [45]. Serum-free light chain concentrations are now routinely measured in clinical practice, particularly in cases of oligosecretory myeloma or suspected light chain amyloidosis, and this facilitates diagnostic evaluation and treatment monitoring.
Early Cases of Multiple Myeloma After Sarah Newbury and Thomas McBean
In 1867, Hermann Weber, a German physician working in London, described a 40-year-old man with mollities ossium and first linked this condition to the pathological finding of tissue deposition of amyloid [46]. The patient suffered severe sternal and lumbar pain, and movement of his head produced pain in his neck and arms. The patient died less than 4 months after the initial onset of pain. Postmortem examination revealed that the sternum was fractured in two places, and had been almost entirely replaced by a grayish red substance that was thought to have the microscopic appearance of a sarcoma. Several round defects in the skull were also filled with the same morbid substance as that found in the sternum, and many of the ribs, several vertebrae, and parts of the pelvis were involved by the same process. The waxy changes of amyloid were found in the kidneys and spleen.
During a meeting of the Pathological Society of London in February 1872, Mr. William Adams exhibited (on behalf of Dr. Thomas Stretch Dowse of Highgate Infirmary) specimens from the body of a 62-year-old woman with “acute rheumatism” characterized by bone pain, fractures, and fever, who died 8 days after admission to the hospital for a humerus fracture. The left femur fractured while the body was being placed on the autopsy table. Lardaceous changes consistent with amyloid were found in the liver and kidneys, and the cancellous portions of the bones had been replaced by a homogeneous, soft, gelatinoid substance. When examined microscopically, the substance filling the hollowed bone was shown to consist of small spherical and oval cells that contained one oval nucleus (rarely two), with a bright nucleus [47].
The term “multiple myeloma” was introduced in 1873 by J. von Rustizky from Kiev, of whom little is known other than that he had once worked in Friedrich von Recklinghausen’s laboratory in Strasbourg. During an autopsy, von Rustizky noted eight separate tumors of the bone marrow in a patient, which he called “multiple myelomas” [48]. The patient, a 47-year-old man, had presented with a gradually enlarging tumor in the right temple. Subsequently, thickening of the sternal manubrium and the seventh rib developed, followed by paraplegia. At autopsy, it was revealed that a fist-sized tumor in the right frontal region extended into the orbit and had produced ophthalmoplegia. Other postmortem findings included an apple-sized tumor in the right fifth rib, a tumor in the left seventh rib that produced a fracture, a tumor of the sternum, a tumor involving the sixth to the eighth thoracic vertebrae (the cause of the paraplegia), and three tumors of the right humerus. Although von Rustizky’s description of the tumor cells in this case is vague, he described round cells with a nucleus located in the periphery near the cell membrane, suggestive of plasma cells. The report did not comment on whether Bence Jones proteinuria was found during life. In Russia and Soviet-influenced regions, multiple myeloma was called “von Rustizky syndrome” for much of the twentieth century.
There were few further publications about the disease until 1889, when Otto Kahler, an internist from Prague working in Vienna, described the case of a 46-year-old physician named Dr. Loos [49]. In July 1879, Dr. Loos developed sudden severe pain in the right upper thorax, which was aggravated by taking a deep breath. Six months later, this pain recurred and became localized to the right third rib, which was tender to pressure. During the next 2 years, intermittent pain aggravated by exercise occurred in the ribs, spinal column, left shoulder, upper arm, and right clavicle. Albuminuria was first noticed in September 1881. Skeletal pain, made worse by movement, continued to occur intermittently. Pallor was noted in 1883 and pneumonia developed in February 1884. In December 1885, Dr. Loos was first seen by Dr. Kahler, who noted anemia, severe kyphosis, and focal tenderness of many bones. When Dr. Loos stood up, his lower ribs touched his anterior iliac crest. Dr. Loos subsequently suffered from recurrent bronchial infections and intermittent hemoptysis. During the following year, Dr. Loos’ kyphosis increased and height decreased monthly, to the point where his chin pressed against the sternum, resulting in skin ulceration. On August 26, 1887, Dr. Loos died, a remarkable 8 years after his initial symptoms.
Kahler’s autopsy report of Dr. Loos described hepatosplenomegaly, but lardaceous change was not mentioned. The ribs were soft and could be broken with minimal effort. Soft gray-reddish masses were noted in the ribs and thoracic vertebrae. Microscopic examination showed large round cells, consistent with myeloma. It is interesting to note that the patient had a high fluid intake and took sodium bicarbonate on a regular basis; this regimen may have helped prevent renal failure. Kahler recognized that the urinary protein obtained from Dr. Loos had the same characteristics that Bence Jones had described. For many years, the eponym “Kahler’s disease” was widely used to describe the condition once known by “mollities ossium with Bence Jones proteinuria” and now called multiple myeloma.
Detailed examination of Dr. Loos’ urine by Karl Hugo Huppert, professor of medicinal chemistry at the German University in Prague, showed that the protein precipitated at 53 °C to 59 °C, cleared with heating to boiling, and then re-precipitated during cooling [50]. The patient excreted 6.7 g of the protein daily, which Huppert noted did not contain albumin.
It is likely that A.L., the 43-year-old engineer that Joseph Coats of Glasgow in 1891 reported as having “multiple sarcoma of bone,” actually had multiple myeloma [51]. The patient developed a large tumor of the sternum 5 years before his final hospitalization and death, and later noticed tumors in the right clavicle, right humerus, and left hip. He experienced back pain radiating to the lower extremities, weakness of his legs, and a pathological fracture of the right humerus. Postmortem examination revealed multiple tumors with involvement of the ribs and vertebral bodies. Microscopic examination showed round or polygonal cells “about 1/2,000 in. diameter” (∼13 μm, a reasonably accurate estimate of the diameter of a neoplastic plasma cell) with oval nuclei constituting more than half the diameter of the cells [51].
An 1897 Italian description of Kahler’s disease by Camillo Bozzolo, a Milanese physician and pathologist working in Torino, resulted in the dual eponym “malattia di Kahler-Bozzolo” gaining currency in Italy in the first part of the twentieth century [52].
Other Cases of Multiple Myeloma in the Nineteenth and Twentieth Centuries
In 1894, James Bryan Herrick and Ludvig Hektoen at Rush Medical College in Chicago reported what is probably the first recognized case of multiple myeloma in the United States, just a few years after the report of Kahler [53]. A 40-year-old woman complained of lumbar pain and a nodule on the lower end of the sternum. At autopsy, there were multiple nodules attached to the sternum, right clavicle, and ribs. The sternum was thickened, irregular, and covered with tumor masses, yet was soft and flexible. Multiple nodules were found on the ribs, which bent readily without cracking. Two of the dorsal vertebral bodies were largely replaced by soft tumor masses, and fungoid masses were seen in the skull. Microscopic examination of these lesions revealed round “lymphoid” cells with large nuclei. Remarkably, Herrick is also credited with the first description of sickle cell anemia, as well as being the first to link angina with acute coronary syndromes [54, 55].
In 1898, Frederick Parkes Weber—honorary physician to the German Hospital in Queen’s Square, London, and son of Dr. Hermann Weber mentioned earlier—reported a case of multiple myeloma, and suggested that radiographs (discovered by Würzburg physicist Wilhelm Conrad Röntgen in 1895) would greatly facilitate the diagnosis of such cases [56]. Weber also concluded in a report of another case that Bence Jones protein was produced by the bone marrow, and that the presence of Bence Jones protein was of “fatal significance” and nearly always indicated that the patient had multiple myeloma [57]. Weber and Lister Institute bacteriologist John Charles Grant Ledingham later suggested that Bence Jones protein derived from cytoplasmic residua of karyolyzed plasma cells [58].
In 1928, C.F. Geschickter and M.M. Copeland from Johns Hopkins reviewed all 425 cases of multiple myeloma reported since 1848 [59]. They called attention to six cardinal features of the disease: multiple tumors of the axial skeleton, pathological fractures, Bence Jones proteinuria, back pain, anemia, and chronic renal disease.
Sternal aspiration of bone marrow during life, described by Mikhail Arinkin in Leningrad in the late 1920s, greatly increased the recognition of multiple myeloma [60]. In 1938, hematologists Nathan Rosenthal and Peter Vogel at Mt. Sinai Hospital in New York reported that only three cases of multiple myeloma had been recognized at the Mt. Sinai Hospital from 1916 to 1935, but that 13 cases were found in the ensuing 2½ years [61]. They attributed this marked increase in recognition to the use of sternal puncture in patients with obscure anemia or skeletal abnormalities, and stated that many cases of myeloma had likely been missed in the past.
In 1947, Edwin (Ned) Bayrd and Frank Heck at Mayo Clinic in Minnesota described 83 patients with histological proof of multiple myeloma seen at their institution through December 1945 [62]. The duration of survival in the Mayo series ranged from 1 to 84 months (median, 15 months). The median survival for myeloma would not increase much beyond this until the introduction of novel agents in the twenty-first century [63].
Plasma Cells, Hyperproteinemia, Recognition of Monoclonality, and Prognosis
The term plasma cell was coined by German anatomist Heinrich Wilhelm Gottfried von Waldeyer-Hartz in 1875, but his description is not characteristic of plasma cells, and it is most likely that he was instead observing tissue mast cells [64]. Plasma cells were described accurately by Spanish neuroscientist Santiago Ramón y Cajal in 1890, during study of syphilitic condylomas. Ramón y Cajal stated that the unstained perinuclear area (“hof,” from a German term meaning yard or court) contained the Golgi apparatus. In 1891, German dermatologist Paul Gerson Unna used the term plasma cell while describing cells seen in the skin of patients with lupus erythematosus [65]. However, it is not known whether he actually saw plasma cells. In 1895, Hungarian pathologist Tamás Marschalkó outlined the essential characteristics of plasma cells, including blocked chromatin, eccentric position of the nucleus, a perinuclear pale area (hof), and a spherical or irregular cytoplasm [66].
In 1900, pathologist J.H. Wright of Johns Hopkins described a 54-year-old man with multiple myeloma, and pointed out that the tumor consisted of plasma cells, proposing the new term “plasma cell myeloma” [67]. Wright emphasized that the neoplasm originated not from red marrow cells collectively but from only one type of cell, the plasma cell. Interestingly, Wright’s patient was probably the first in whom radiographs revealed changes in the ribs, thus contributing to the diagnosis.
Although in 1917 Victor C. Jacobson of the Peter Bent Brigham Hospital in Boston reported Bence Jones protein in the serum in a patient with chronic nephritis [68], it was not until 1928 that W.A. Perlzweig and colleagues from Duke University reported hyperproteinemia when they described a patient with multiple myeloma who had 9–11 g of globulin in his serum [69]. The patient also had Bence Jones proteinuria and probably a small amount of Bence Jones protein in the plasma. Perlzweig and co-workers noted that it was almost impossible to obtain serum from the clotted blood because the clot failed to retract, even on prolonged centrifugation.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


