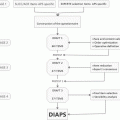
Years
Advances
1
1906–1962
Identifying an antigen, associating the antibody with systemic lupus erythematosus
2
1952–1980
Defining lupus anticoagulant, understanding a mechanism
3
1969–1985
Defining and measuring anticardiolipin
4
1963–1985
Defining a syndrome: thrombosis and pregnancy
5
1985–1989
Primary APS
6
1987–1991
A sub-syndrome: catastrophic APS
7
1990–1994
Animal models
8
1990–1999
β2-Glycoprotein I
9
1996–2016
Criteria and international collaboration
1906–1962: Identifying an Antigen, Associating the Antibody with Systemic Lupus Erythematosus
In 1905 August Paul von Wassermann, recognizing the spirochete, Treponema pallidum, to be the cause of syphilis, used complement fixation, an immunological test available in his era, to identify an antibody that binds the surface of the spirochete. Thus, he created a diagnostic test for syphilis that has lasted to this day [4]. It soon became apparent that Wassermann’s antibody is also found in patients who do not have syphilis, a finding that led to the concept of biological false-positive test for syphilis (BFP). By the 1940s physicians recognized that the BFP is a signal of autoimmunity, since many patients with BFP also have systemic lupus erythematosus (SLE) or a related autoimmune disease [5]. Over the next two decades a team of physicians at the Johns Hopkins Hospital in Baltimore, Maryland (A. McGee Harvey, Lawrence E. Shulman, J.E. Moore, Philip Tumulty, and C.E. Conley) published a series of papers that described the relationship between BFP and SLE [6–9].
In 1941 Mary C. Pangborn, from the Division of Laboratories and Research, New York State Department of Health in Albany, New York, purified the syphilis antigen responsible for a positive Wassermann test. Because the antigen is a phospholipid extracted from beef hearts, she named it cardiolipin [10]. Forty years later this phospholipid reappeared at the center of the description of APS.
1952–1980: Defining Lupus Anticoagulant, Understanding a Mechanism
Independent discoveries by hematologists in the 1950s called attention to an association between BFP and a blood-borne inhibitor of coagulation, now termed lupus anticoagulant (LA). Lupus anticoagulant was initially thought to be a cause of hemorrhage [11, 12]. However, in 1959, a Mexican group led by L. Sánchez-Medal suggested that LA might be pro-thrombotic [13]. In 1963 E.J. “Walt” Bowie from the United States [14], and in 1967 Donato Alarcon-Segovia from Mexico [15], published clinical and mechanistic papers that more firmly established LA as a pro-thrombotic factor. In the 1970s an American group led by Samuel I. Rapaport demonstrated that the LA is an immunoglobulin of IgG or IgM isotype and its activity requires phospholipid [16, 17]. In 1980 Sandor Shapiro [18] (a mentor of Vittorio Pengo [19, 20]), in Philadelphia, demonstrated in vitro and presumably in vivo that a molecule responsible for LA activity, which he isolated from a patient with Waldenstrom’s macroglobulinemia, is an IgM antibody that, in an Ouchterlony precipitation assay, directly binds phospholipids.
Carl Alving, at the Walter Reed Institute in Washington, D.C., demonstrated in 1969 that lipids are immunogenic [21]. This finding, published in the biochemistry literature and unassociated with clinical concepts, lays fallow for a decade until, in 1980, Moshe Smolarsky, crediting Alving, described a radioimmunoassay that can identify antibodies to lipids [22].
1969–1985: Defining and Measuring Anticardiolipin
The association of BFP with SLE created a clinical conundrum because it is not a practical test. Uncommon in a community and more often true than falsely positive, to screen for syphilis is a poor way to screen for autoimmune disease. The test for LA is impractical, since it is also uncommonly present; as initially performed, it requires fresh plasma and an available, capable laboratory. Furthermore, the test for LA is not easily standardized. Thus a simpler way of identifying this phenomenon was required.
In 1983, E. Nigel Harris [23], working with Graham R. V. Hughes, used a radioimmunoassay, followed very shortly by a similar, more convenient, Enzyme-linked Immunosorbent Assay (ELISA) [24], to identify antibodies to cardiolipin. Use of this test markedly simplified the identification of aPL in patients with SLE. Through a series of conferences organized by Hughes, Harris, and Azzudin Gharavi, this test was standardized and internationalized, and the term antiphospholipid antibody (or anticardiolipin antibody) was first used in a clinical context [25]. The Hughes group generously shared their assay with others, allowing its widespread use and confirmation throughout the world. The first international meeting on aPL and its associated syndrome, APS (see below), organized by Hughes, took place in 1984 in London.
1963–1985: Defining a Syndrome (Thrombosis and Pregnancy)
The links between LA, SLE, and thrombosis have been known since the early 1960s. The association of LA with pregnancy loss was first published in 1975 by Inga Marie Nilsson [26], in English, in a not widely read Scandinavian journal. The same association was noted and published in French by J. P. Soulier and Marie-Claire Boffa in 1980 [27]. These papers were not widely cited until the radioimmunoassay and ELISA tests for anticardiolipin became available in 1983. Initially, aCL was thought to be a more rapid, more reliable, test for LA, that is, equivalent to LA, and to impart the same clinical features. Clinical studies on the frequent occurrence of aCL in SLE patients rapidly led to the recognition that both thrombosis and pregnancy loss occur primarily in those who carry aCL/LA. This special form of SLE was soon called anticardiolipin syndrome , or lupus anticoagulant syndrome, or APS (the preferred term) [28].
The rapid use of the aCL ELISA worldwide led to an explosion of clinical papers that further defined the classical (thrombosis and pregnancy loss) manifestations of APS as well as nonclassical manifestations, like livedo reticularis, heart valve abnormality, thrombocytopenia, and cognitive dysfunction. Parameters of APS-associated pregnancy complication—early preeclampsia, growth restriction, thrombocytopenia , and HELLP syndrome (hemolysis, elevated liver enzymes, low platelets)—were described in more detail [29, 30].
1985–1989: Primary Antiphospholipid Syndrome
Within a few years many investigators, beginning with Ronald A. Asherson from Hughes’ group in England in 1985 [31], described patients with APS who did not have diagnosable SLE. In 1987 Harris, in an editorial in which he described the essential features of APS, suggested that APS is an independent entity distinct from SLE. He called it the “syndrome of the black swan” [32]. 1989 brought several more reports from the Hughes group in England (Asherson [33] and Charles G. Mackworth-Young [34]) and Mexico (Alarcón-Segovia [35]) that dissociated APS from SLE and distinguished “primary” antiphospholipid syndrome (PAPS) , in which SLE is not present, from “secondary” antiphospholipid syndrome (sAPS), in which APS accompanies SLE or a related illness.
1987–1991: A Sub-syndrome: Catastrophic Antiphospholipid Syndrome
In 1987 Asherson described a devastating complication of APS, multiple nearly simultaneous thromboses, now called catastrophic APS or CAPS [36]. Although some refer to CAPS as Asherson’s syndrome , A. M. Harvey may have described CAPS in 1954 [9]. Two other papers published in 1987 also described CAPS [37, 38], as did a third paper in 1988 [39]. Asherson also described a signal manifestation of CAPS, adrenal infarction, in 1989 [40], the same year in which Alarcon-Segovia discussed the non-inflammatory vasculopathic pathology of CAPS [41]. R. D. Collins may have been the first to use the term “catastrophic, ” also in 1989 [42], and Stewart Greisman presented several cases with accompanying histologic pathology in 1991 [43]
1990–1994: Animal Models
Beginning in the 1990s, animal models enhanced our understanding of the mechanisms of APS. D. Ware Branch published a pregnancy loss model in 1990 [44], and Miri Blank and Yehuda Shoenfeld offered a similar model in 1991 [45]. Silvia Pierangeli described an animal model for thrombosis in 1994 [46]. The pregnancy and the thrombosis models have been widely and successfully exploited for mechanistic and treatment studies since that time. They remain in use today [47–49].
1990–1999: β2-Glycoprotein I
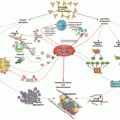
In vitro the ELISA for aCL behaves aberrantly, becoming nonlinear when the tested serum sample is diluted with albumin or fetal calf serum instead of adult human or animal serum. This anomaly led investigators in Maastricht, Sydney, Sapporo, and Paris nearly simultaneously to identify a cofactor necessary for immunoglobulin binding to phospholipid [50–54]. This cofactor is now known to be β2-glycoprotein I (apolipoprotein H, β2GPI); aPLs are now thought to be primarily antibodies to β2GPI and only secondarily, through β2GPI, are they directed against phospholipids. Extensive molecular biological studies have defined the molecular and tertiary structures of β2GPI, its five domains, its binding sites, and its antigenic sites.
Many papers on the biology of aPL and APS [55–57] focus on the molecular biology of β2GPI, its antibodies and its receptors, its role in complement activation, endothelial and platelet activation, cytokines, pro- and antithrombotic proteins, placental biology, genomics and microbiomics, and other areas. The induction of aPL or antibody to β2GPI by infectious agents, from viral (Epstein-Barr virus) to mycobacterial (leprosy), is a topic under active exploration. Alternatively, aPL may cross-react with antibodies to infectious agents. Treatments, including inhibitors of peptides on β2GPI, complement, receptors, and B cells, competing antibodies, antiplatelet agents , and new and old anticoagulants, all target novel pathogenic pathways and mechanisms.
1996–2016: Criteria and International Collaboration
Largely through the efforts of the Hughes group in London, the field of APS has been collaborative since the beginning. International meetings have taken place every 2 or 3 years since 1984, the conference in Istanbul (relocated to North Cyprus) (www.apsistanbul2016.org) in 2016 being the Fifteenth. (Pierangeli and Harris summarized the history of these meetings through the Thirteenth [58].) The European Forum began in 1996 [59]; APS Alliance for Clinical Trials and International Networking (APS-ACTION) , a group dedicated to clinical trials, began in 2010 [60]. International consensus committees have devised clinical and laboratory criteria for classification, first in Sapporo in 1998 [61], with revision in Sydney in 2004 [62], and further efforts to develop new classification criteria under the auspices of the Fifteenth Congress [63]. Major hospital clinics focused on APS now exist in most areas of the world.
The Future
This history necessarily focuses on past events. Many important clinical questions—clear predictive ability, fully satisfactory treatment, possibly cures—are not yet available. The following questions require answers: