Fig. 2.1
Transitional meningioma showing characteristic concentric whorls and basophilic psammoma bodies. Magnification ×200
Meningiomas located in the cerebellopontine angle or in relation to cranial/spinal nerve roots raise a differential diagnosis of Schwannoma (neurilemmoma). Histologic differentiation can often be done readily based on light microscopic and immunostaining features. Schwannomas typically show positivity for S-100 protein and negative staining for epithelial membrane antigen. In contrast, meningothelial cells are positive for epithelial membrane antigen and claudin-1 among others and are rarely positive for S-100 protein. Furthermore, electron microscopy demonstrates desmosome junctions, cytoplasmic interdigitations, and a lack of basal lamina in the tumor cells as is characteristic of meningioma, while the presence of a basal lamina and a lack of true desmosome junctions are consistent with Schwann cell differentiation.
A demonstrable invasion of underlying brain is indicative of an aggressive meningioma and is often accompanied by histological features of an atypical or malignant meningioma.
2.1.1.1 Atypical Meningioma (WHO Grade II)
Atypical meningioma represents subset of meningioma with slightly higher risk of recurrence when compared with benign meningiomas (29–39% vs. 7–20%). Such tumors are characterized by increased mitotic rate (>4/10 hpf in the area of highest mitotic activity) or at least three of the following histologic findings: hypercellularity, small cell change, prominent nucleoli, sheet-like growth, or foci of spontaneous or geographic necrosis (i.e., not induced by therapeutic embolization). Proliferation index (MIB-1 index) is usually greater than 5% (mean 2.1% vs. mean for benign meningiomas of 0.7%).
Meningiomas with tongue-like invasion of underlying brain and chordoid or clear cell features are clinically aggressive and classified as atypical meningiomas (WHO grade II).
2.1.1.2 Anaplastic (Malignant) Meningioma
- 1.
Papillary meningioma
Papillary meningiomas are by definition malignant meningiomas and the presence of a distinct papillary pattern is the hallmark of this subset of meningiomas (Fig. 2.2). They represent a WHO grade III. Papillary meningiomas tend to occur more commonly in children. Papillary features in a dural-based lesion should raise the differential diagnosis of a metastatic papillary adenocarcinoma.
- 2.
Malignant (non-papillary) meningioma
The diagnosis of anaplastic or malignant meningioma in a non-papillary tumor requires the presence of overt or frank anaplasia and/or metastases. The more classic lesions are usually characterized by increased cellularity, frequent mitotic figures (>20 mitoses/10 hpf), and conspicuous necrosis. The proliferation index (MIB-1 index) is usually greater than 10% (mean = 11%). Rhabdoid meningioma is an aggressive variant of meningioma and is classified also as a WHO Grade III.
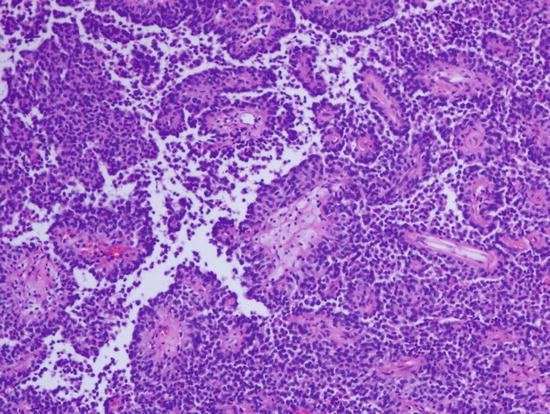
Fig. 2.2
Papillary growth pattern is a characteristic feature of papillary meningioma. Magnification ×100
Postsurgical recurrence is a common feature of meningiomas and is seen in up to 20% of benign meningiomas after 20 years of follow-up. Indicators of possible recurrence (apart from incomplete resection) are histological subtype and tumor grade. Anaplastic meningiomas have the highest recurrence rate of about 50–78%.
2.1.2 Mesenchymal, Non-meningothelial Tumors
This group of tumors share similar histological features with their counterparts in peripheral soft tissues. They include:
- 1.
Adipose tissue tumors, such as lipomas [fibrolipomatous hamartomas, angiolipomas, and epidural lipomatosis (related to chronic corticosteroid administration)] and intracranial liposarcoma. Lipomas occur most often in the midline, typically located in the corpus callosum, cerebellar vermis, or mammillary bodies. They may also occur as part of a complex malformation. Mature lipocytes have been described as part of a low-grade cerebellar tumor called liponeurocytoma that is composed of intermixed mature lipocytes and neurocytic cells.
- 2.
Fibrohistiocytic tumors, including benign and malignant fibrous histiocytoma.
- 3.
Fibrous tumors including solitary fibrous tumor, hypertrophic intracranial pachymeningitis, and fibrosarcoma.
- 4.
Muscle forming tumors, such as leiomyoma, intracranial leiomyosarcomas, rhabdomyoma, embryonal rhabdomyosarcoma, and malignant ectomesenchymoma.
- 5.
Osteocartilaginous tumors, including chondroma, osteoma and osteochondroma, mesenchymal chondrosarcoma, and osteosarcoma.
- 6.
Vascular tumors, such as hemangiomas, epithelioid hemangioendotheliomas, angiosarcoma, and Kaposi’s sarcoma.
- 7.
Tumors of undefined histogenesis, such as hemangiopericytoma, capillary hemangioblastoma, and meningeal sarcoma/sarcomatosis.
- 8.
Melanocytic tumors.
Since many of these tumors are curiosities in the CNS, only the relatively more frequent tumors will be discussed further. The histological features of the other tumors are similar to those of their soft tissue counterparts and are well discussed in the soft tissue sections of other standard textbooks.
2.1.2.1 Solitary Fibrous Tumor/Hemangiopericytoma
Historically, this tumor has been referred to by various obsolete names such as “angioblastic meningioma” and hemangiopericytic variant of meningioma reflecting a lack of understanding of its true histogenesis. Hemangiopericytoma accounts for only 0.4% of all brain tumors and is a rare tumor in children. It is usually dural based and is rarely intraparenchymal. Clinical presentation is similar to that of meningioma. The cellular variant of solitary fibrous tumor (SFT) shows histologic and immunohistochemical overlap with that of hemangiopericytoma with nuclear immunopositivity for STAT6 and both are regarded as same entities.
Grossly, the tumor is firm, well demarcated, globoid, slightly lobulated, and can bleed profusely during surgical removal. Histologically, it is characterized by monotonous sheets of oval to elongated nuclei, variable degrees of nuclear atypia, and prominent mitotic activity (Fig. 2.3). “Staghorn” vessels of variable prominence are also apparent. Invasion of underlying brain can occur. Since many soft tissue tumors can have hemangiopericytomatous-like areas, immunostaining and electron microscopy, if available, are important aids in ensuring accurate diagnosis. Hemangiopericytomas show an identical immunohistochemical profile as its soft tissue counterpart (now regarded as solitary fibrous tumor) with diffuse positivity for vimentin, CD99, and bcl-2, and more variable positivity for Leu-7, CD34, and factor XIIIa in individual tumor cells. CD34 positivity is generally patchy as opposed to the diffuse positivity typical of solitary fibrous tumor. Unlike the strong diffuse positivity for EMA and claudin-1 displayed by meningiomas, staining is typically patchy and weak for these antibodies in hemangiopericytoma. They are negative for S-100, CD31, and progesterone receptor. Actin, desmin, and cytokeratin (CAM5.2) staining is rare. p53 immunoreactivity may be seen in about one-half of HPC, whereas this is not generally a feature of meningioma or SFT.
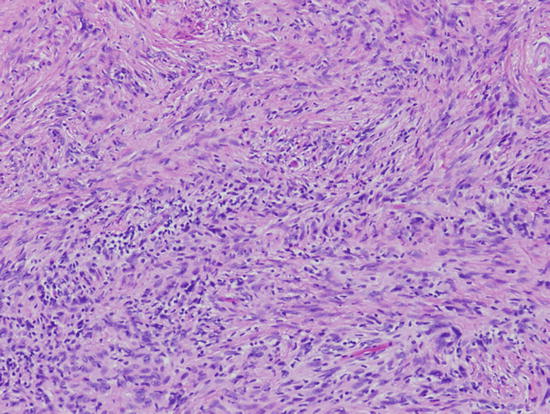
Fig. 2.3
Cellular and fibrosing spindle cell proliferation of a cellular solitary fibrous tumor/hemangiopericytoma. Magnification ×200
The proliferation index (MIB-1 index) varies widely with median values ranging between 5 and 10%. Hemangiopericytomas exhibit a high 15-year local recurrence rate of 85–91% and metastases are also seen in about 65% of cases when followed for over 15 years.
2.1.2.2 Capillary Hemangioblastoma
Capillary hemangioblastoma is a WHO grade I vasoformative tumor of uncertain histogenesis which commonly occurs in the cerebellum. Spinal and brain stem lesions also occur, but less frequently, and supratentorial lesions are very rare. About 25% of cases occur in the setting of Von Hippel–Lindau disease. Von Hippel–Lindau disease-associated tumors occur in younger patients while the non-Von Hippel–Lindau disease-associated tumors occur in adults. Von Hippel–Lindau disease is a neurocutaneous syndrome associated with retinal hemangioblastoma, pheochromocytoma, renal cell carcinoma, and visceral (liver and pancreas) cysts. Hemangioblastomas are slow growing tumors often presenting with features of raised intracranial pressure secondary to the blockage of CSF flow. There is often an accompanying secondary polycythemia due to erythropoietin production by the neoplastic stromal cells.
Grossly, capillary hemangioblastoma consists of well-circumscribed red nodules often in the wall of large cysts. Histologically, the tumors are composed of large vacuolated stromal cells and a rich capillary network. The stromal cells show immunopositivity for vimentin, but the tumors do not show evidence of immunoreactivity for glial fibrillary acidic protein or for endothelial markers such as CD34 or Von-Willebrand factor. The “clear cell” appearance of the stromal cells can be confused with metastatic renal cell carcinoma. Rosenthal fibers can be seen in the wall of the cysts. The slow growth of capillary hemangioblastoma is consistent with a low proliferation index (MIB-1 index <1%). The stromal cells are consistently positive for vimentin, alpha inhibin, D2-40, and aquaporin, while variably positive for S-100, NCAM, NSE, erythropoietin, EGFR, VEGF, alpha-1-antitrypsin, and antichymotrypsin. Progesterone receptor and Factor XIIIa positivity have also been reported in a high percentage of cases. They may be focally positive for GFAP, keratin, EMA, or desmin, while specific neuronal (neurofilament, synaptophysin, and chromogranin) and endothelial cell (CD31, CD34, and VWF) markers are typically negative.
2.1.2.3 Meningeal Sarcomatosis
Meningeal sarcomatosis is a diffuse leptomeningeal sarcomatous tumor lacking the distinct circumscribed nature that is characteristic of most meningeal tumors. It is composed of poorly differentiated spindle cells characteristic of undifferentiated sarcomas. Apart from immunostaining for vimentin, the tumor cells are negative for all neuroglial markers.
2.1.2.4 Mesenchymal Chondrosarcoma
The most common extraosseous site for mesenchymal chondrosarcoma is the CNS. Mesenchymal chondrosarcoma has a distinct small cell tumor component interrupted by islands of atypical hyaline cartilage (Fig. 2.4). The small cell component shares histological features with hemangiopericytoma and can be confused with it in small biopsies that lack the characteristic chondroid component.
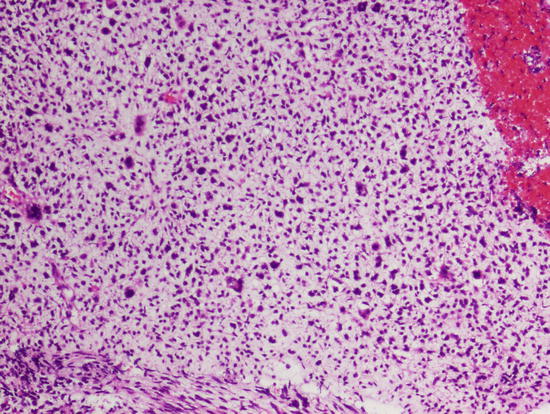
Fig. 2.4
Malignant chondromyxoid proliferation of mesenchymal chondrosarcoma. Note the nuclear pleomorphism. Magnification ×200
2.1.2.5 Melanocytic Lesions
Primary melanocytic lesions of the CNS are relatively uncommon, accounting for only 0.06–0.1% of brain tumors. Melanocytic lesions are more common in Caucasians and arise from melanocytes of the leptomeninges. There are three possible forms of CNS melanocytic lesions: (1) diffuse melanocytosis, (2) melanocytoma, and (3) primary leptomeningeal malignant melanoma.
- 1.
Diffuse melanocytosis usually presents in childhood with seizures, behavioral disturbances, and hydrocephalus. Histologically, the lesion shows a diffuse proliferation of uniform nevoid polygonal cells within the leptomeninges.
- 2.
Melanocytomas present as mass lesions composed of a monomorphic population of spindle, fusiform, epithelioid, or polyhedral cells arranged in whorls, sheets, nests, or interlacing bundles of storiform configuration with infrequent mitotic figures. Melanin can be present. The tumors are S-100 protein positive and in our experience are usually negative for the melanoma antigen markers, HMB-45, and Melan A.
- 3.
Primary meningeal malignant melanomas also present as mass lesions. They occur classically in the setting of neurocutaneous melanosis, such as in the autosomal dominant Touraine syndrome and in patients with the congenital nevus of Ota. The tumors have similar aggressive behavior as seen in cutaneous melanoma and are characterized histologically by marked pleomorphism, high mitotic activity, necrosis, hemorrhage, and invasion of underlying brain. The diagnosis of a primary CNS melanoma can only be made after a metastatic melanoma has been excluded. As in cutaneous melanoma, the tumor cells are S-100 protein, HMB-45 and Melan A positive while being negative for glial fibrillary acidic protein (GFAP), neurofilament protein (NFP), epithelial membrane antigen (EMA), and cytokeratin. Electron microscopy shows the presence of melanosomes.
2.2 Astrocytic Tumors
Pilocytic astrocytoma (PA) represents a slow growing, often circumscribed fibrillary astrocytoma with a distinctly good prognosis warranting its designation as a WHO grade I. It accounts for 20–25% of all childhood brain tumors occurring frequently as a cystic cerebellar tumor with a mural nodule. Similar tumors may involve the optic nerve, presenting as optic nerve glioma in children and frequently in individuals with neurofibromatosis type 1 (NF1). Supratentorial intra-axial tumors are common in the temporal lobe and may also involve the hypothalamus or thalamus. The characteristic cells are the glial fibrillary acidic protein-positive piloid (elongated or “hair-like”) astrocytes which are arranged in a biphasic pattern of cellular fibrillary astrocytes with intervening loose microcystic areas (Fig. 2.5). Rosenthal fibers and eosinophilic granular bodies (EGBs) are frequently present and the abundance of Rosenthal fibers is a helpful diagnostic feature in frozen sections or crush preps made during intraoperative consultation. Unlike the classic diffuse infiltrating astrocytoma, the presence of vascular proliferation and slight pleomorphism does not imply an aggressive high histologic grade. However, in the rare case, the presence of frequent mitotic figures, a significantly increased cellularity, and necrosis justifies the diagnosis of a pilocytic astrocytoma with anaplastic features. Tandem duplication at 7q34 with BRAF-KIAA1549 duplication/fusion rearrangement is seen in 70% of PA. BRAFV600E mutation is seen in 10% of extra-cerebellar PA, particularly diencephalic tumors. Rare co-occurrence of both events has been seen.
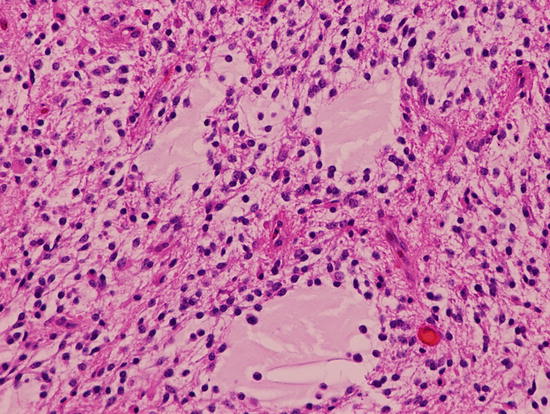
Fig. 2.5
Pilocytic astrocytoma with biphenotypic microcystic and compact cellular components. Eosinophilic Rosenthal fiber is present. Magnification ×200
The pilomyxoid astrocytoma (PMA), WHO grade II, shares histologic features with the PA. It shows additional features including a monomorphous population of bipolar cells with delicate elongated “piloid” processes in an abundant myxoid matrix. The cell processes frequently radiate from vessels with a pseudorosette pattern. In its pure form, Rosenthal fibers and EGBs are absent. Mitoses may be seen. Maturing PMAs show intermediate histologic features of both PA and PMA.
BRAF-KIAA1549 duplication/fusion rearrangement is seen in 60% of PMA.
Pleomorphic xanthoastrocytoma (PXA), WHO Grade II, is a tumor with distinct radiopathologic features including a superficial meningocerebral location, common location in the temporal lobe, and frequent association with a long-standing history of seizures. Pleomorphic xanthoastrocytoma accounts for <1% of all astrocytic neoplasms but 2/3 of cases occur in individuals below the age of 18 years. Histologically, it exhibits significant pleomorphism with many atypical giant cells and astrocytic cells with slightly prominent nucleoli. Mitotic activity and necrosis are conspicuously absent in the typical pleomorphic xanthoastrocytoma. There is a variably discernible population of foamy (xanthomatous) cells and focal lymphocytic and plasma cell infiltrate in a background of reticulin-positive desmoplasia. The astrocytic origin of this tumor has come into question because of the demonstration of neuronal markers in subpopulations of tumor cells. In addition, the morphologic pattern seen in pleomorphic xanthoastrocytoma may form the glioma component of a ganglioglioma. BRAFV600E mutation is a frequent event seen in 75% of PXA.
Pleomorphic xanthoastrocytoma with anaplastic features is a variant which may represent WHO Grade III. These are often seen as part of tumor progression in recurrent lesions. They show features indicative of aggressive behavior such as increased mitotic activity [> 5 mitosis per 10 high power fields], necrosis, and endothelial proliferation.
Diffuse astrocytomas are infiltrating fibrillary neoplasms with varying degrees of differentiation and tumor grade. They account for 50% of primary brain tumors. Astrocytic tumors characteristically have infiltrative margins and range from the low grade diffuse astrocytoma which is WHO grade II (peak age of 30–39 years) to the anaplastic (malignant) astrocytoma which is WHO grade III (peak age of 40–49 years), and the most aggressive glioblastoma which is WHO grade IV (peak age of 50–69 years). Approximately 10% of all glioblastomas occur within the first two decades of life. Infiltrative astrocytomas are second to pilocytic astrocytoma in frequency in the pediatric age group.
Diffuse infiltrating astrocytoma, WHO grade II, or well-differentiated (low grade) astrocytoma is most common in the cerebral white matter and accounts for 20% of primary brain tumors. Low grade astrocytomas are composed of a relatively uniform population of proliferating astrocytes in a fibrillary matrix (Fig. 2.6). Mitotic figures are rare. In contrast to reactive gliosis, diffuse astrocytomas have infiltrative poorly defined margins and can have microcystic degeneration. The tumor cells show slight atypia, an important criterion for distinguishing them from reactive gliosis. The tumor cells can have plump eosinophilic cytoplasm (gemistocytes) and are often a minor component of most diffuse astrocytomas. Tumors composed of greater than 20% of gemistocytic tumor cells are called gemistocytic astrocytoma. The astrocytic tumor cells show cytoplasmic positivity for glial fibrillary acidic protein.
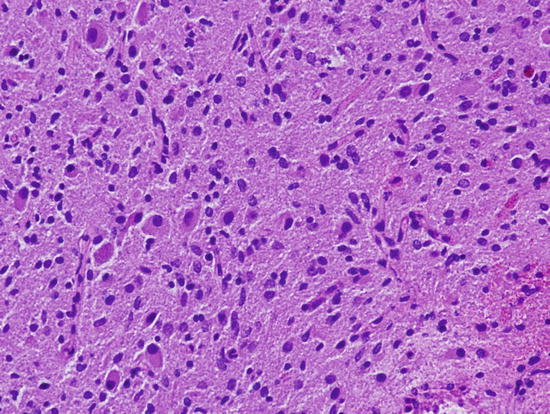
Fig. 2.6
Low grade diffuse infiltrating astrocytoma with mild increase in cellularity, astrocytic differentiation, and rare mitotic figures. Magnification ×200
2.2.1 Anaplastic (Malignant) Astrocytoma
Anaplastic astrocytoma represents a high grade diffuse fibrillary astrocytoma classified as WHO grade III. It has a similar distribution to low grade astrocytoma but is characterized by a greater degree of cellularity and pleomorphism. Mitotic figures are readily discernible (Fig. 2.15).
2.2.2 Glioblastoma
Glioblastoma is the most common glioma, accounting for 50% of all gliomas. It is clinically the most aggressive glioma and represents an extreme expression of astrocytic anaplasia. Glioblastoma occurs frequently as a white matter lesion, is diffusely infiltrative, and can cross the midline by involving the corpus callosum to produce the radiologic “butterfly pattern.” Glioblastoma is characteristically grossly hemorrhagic and necrotic. Histologically, it shows marked cellular pleomorphism, frequent mitotic figures, tumor giant cells, endothelial proliferation, and necrosis with or without pseudo-palisading (Fig. 2.7). A high proliferation index and immunopositivity for p53 protein is a common feature of glioblastoma.
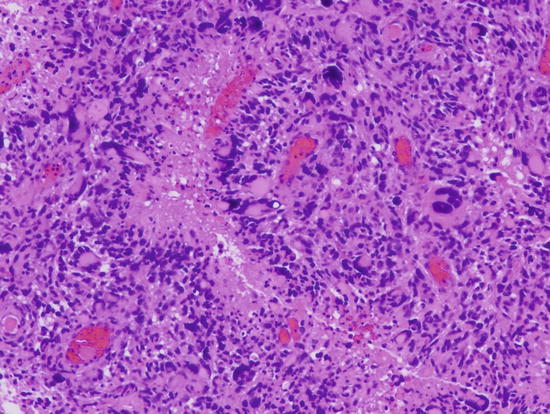
Fig. 2.7
Glioblastoma with increased cellularity, marked pleomorphism and pseudo-palisaded necrosis. Vascular endothelial proliferation is also present (not shown). Magnification ×200
A predominant population of highly proliferative small cells can be seen in the variant form called small cell glioblastoma. The predominance of such cells often raises a differential diagnosis of “small blue cell tumors.” Although cellular pleomorphism is an inherent histologic feature of glioblastoma, an extremely pleomorphic variant with florid multinucleated giant cells constitutes the giant cell glioblastoma. A spindle mesenchymal sarcomatous transformation can also be seen in a variant referred to as the gliosarcoma which must be differentiated from the secondary diffuse spindle (fibroblastic) cell proliferation that can accompany the invasion of the leptomeninges by glioblastoma cells. The giant cell glioblastoma has a slightly better prognosis than the classic glioblastoma while the gliosarcoma has no significantly different outcome from the classic glioblastoma.
Mutation of the p53 gene has been shown to be especially common in the evolution of low grade astrocytoma and in the progression from low grade astrocytoma to high grade astrocytoma. Amplification of mdm2 gene which provides an alternative pathway for p53 inactivation only occurs in a minor subset of glioblastomas. The subset of glioblastomas arising secondary to p53 mutation or inactivation has been designated as secondary glioblastoma. In contrast, de novo or primary glioblastomas are less likely to have mutations of the p53 gene and are more likely to have amplification of the epidermal growth factor receptor gene. Therefore, there seem to be at least two largely separate operant pathways in the biologic evolution of the classic types of glioblastoma.
Additional genetic events in the development of high grade gliomas include epidermal growth factor (EGF) and transforming growth factor-α (TGF-α) expression providing a loop for autocrine stimulation. Fibroblast growth factor and vascular endothelial growth factor overexpression probably plays a major role in the development of angiogenesis, a critical element in the transformation of low grade astrocytoma to glioblastoma multiforme. Other reported genetic events in astrocytoma progression include loss of the deleted in colon carcinoma (DCC) gene, loss of heterozygosity (LOH) for chromosome 10q23.3 (PTEN gene locus) which is mutated in 30–49% of high grade gliomas, total loss of chromosome 10, loss at chromosome 19q13.3, and loss of chromosome 22q. The giant cell glioblastoma does not appear to share these molecular pathways thus suggesting that it is a distinctly different biologic entity, a feature consistent with its differing clinical aggressiveness.
Based on a combination of studies including determination of epigenetic/methylation status as well as gene copy number and expression profiles, glioblastomas can be divided into distinct molecular and prognostic subgroups:
- 1.
H3F3a K27M and G34R mutated subgroups which represent a predominant group among pediatric glioblastomas. These are usually negative for IDH1 mutations, frequently show TP53 mutations, and generally display widespread genomic hypomethylation.
The K27M subgroup is more frequent in children and is associated with midline tumors (DIPG and GBM of thalamus and spinal cord). The G34R subgroup is most frequent in adolescents with tumors of cerebral hemispheric location.
- 2.
IDH1 mutated tumors represent a minor proportion of pediatric glioblastomas, display global genomic hypermethylation, and frequently harbor TP53 mutations. This group includes older children/young adults with tumors of cerebral hemispheric location.
- 3.
PDGFRA amplified tumors are associated with a proneural gene expression profile. This group has a wide age distribution, with a proportion occurring in pediatric tumors with cerebral hemispheric location.
- 4.
Mesenchymal subgroup tumors exhibiting a mesenchymal gene expression signature and no defining gene copy number alterations or point mutations. This group also has a wide age distribution, with a proportion occurring in pediatric tumors of cerebral hemispheric location.
- 5.
“Classic” subgroup is limited to GBM arising in older adults, and this group shows high frequency of chromosome 10 loss, homozygous deletion of CDKN2A, and EGFR amplification.
Diffuse infiltrating pontine glioma (DIPG) represents a diffusely infiltrative glioma involving the basis pontis which may histologically be low grade or high grade. It accounts for 10% of childhood brain tumors and is composed of fibrillary astrocytes. The diagnosis is often based on its classic neuroradiologic presentation with no attempts at resection or biopsy. Treatment usually involves radiotherapy, but there is an attendant high propensity of surviving tumor cells to dedifferentiate to a more anaplastic histology including glioblastoma.
A number of genetic events have been reported in DIPGs including: (1) K27M mutations in H3F3A (H3.3) or HIST1H3B (H3.1) which are extremely common and present in nearly 80% of DIPG. (2) Gain of function ACVR1 mutations found exclusively in DIPG (up to 30% of cases), whereas FGFR1 mutations or fusions are seen in thalamic HGAs. (3) Global DNA hypomethylation is an epigenetic alteration frequently encountered in DIPG, and may be detectable as decreased tumor staining by H3K27me3-specific immunohistochemistry. (4) TP53 (75%) and ATRX (10%) mutations may be encountered in subpopulations of DIPG, with ATRX mutations being more frequent in tumors arising in older children. PIL3CA mutations and PDGF-a gains/amplifications have been reported. Subgroups with frequent loss of 17p and 14q and others with lack of IDH1/2 mutations or CDKN2a/CDKN2B deletions have also been reported.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


