Fig. 7.1
Peripheral blood film showing typical CLL cells and a smear cell (1,000× magnification)
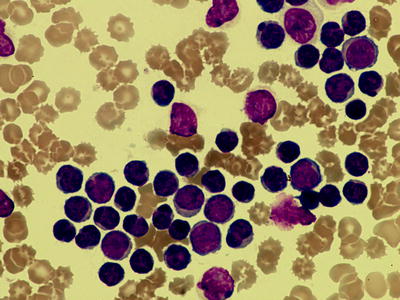
Fig. 7.2
Peripheral blood film showing prolymphocytes along with typical CLL cells (1,000× magnification)
In CLL/PL as defined by the FAB classification prolymphocytes comprise between 10 and 55 % of cells. It is recognized that such cases are characterized by a higher incidence of adverse molecular and cellular features, such as trisomy 12, proliferation rate and unmutated V region of the immunoglobulin (Ig) gene [36–39]. It is also recognized that the number of prolymphocytes can increase during the course of disease. With the advent of modern immunophenotyping and genotyping characterization these morphological sub-classifications have become less relevant. It has been clear over recent years that cases comprising almost entirely of prolymphocytes rarely have the immunophenotypic or genotypic features of CLL and B-PLL is recognized as a distinct entity.
Histopathology
Bone marrow is the commonest tissue available for histological examination and immunohistochemistry in CLL. Although bone marrow aspirate and biopsy are generally not required to make the diagnosis of CLL, if a marrow is done the aspirate smear must show more than 30 % of all nucleated cells to be lymphoid for the diagnosis of CLL. The pattern of marrow infiltration was previously thought to have prognostic significance, but with the advent of newer prognostic markers it has become less relevant [40]. Even though marrow is not essential for the diagnosis of CLL it is usually recommended before initiating treatment in CLL. The trephine biopsy is a better guide to the overall level of infiltration compared to a bone marrow aspiration. The trephine biopsy core should preferably be a minimum of 1.6 cm, ideally 2 cm, in length taken from the posterior iliac crest [41, 42]. It is valuable particularly in the context of cytopenias both at diagnosis and following treatment as it allows the distinction between marrow failure due to disease, autoimmunity, hypersplenism or treatment-related effects. In around 25 % of patients autoimmune haemolytic anaemia can occur, where the bone marrow will show erythroid hyperplasia [43, 44]. Marrow fibrosis can occur but is rare [45]. Pure red cell aplasia can occur as an autoimmune complication in about 1 % of patients and rarely autoimmune granulocytopenia can also occur [46].
The morphology of lymphocytes in the aspirate is similar to the pattern observed in peripheral blood (Fig. 7.3). Cytology is, however, better appreciated on peripheral blood smears. Medullary spaces can be completely replaced or can be infiltrated in a patchy manner. Bone marrow infiltration in CLL can be interstitial, nodular, diffuse or mixed pattern. In the diffuse growth pattern the marrow is hypercellular and normal haematopoietic cells and fat spaces are replaced by CLL cells (Figs. 7.4 and 7.5). In nodular pattern there is discrete, non-paratrabecular aggregates of small lymphocytes scattered throughout the marrow space. Benign lymphoid aggregates that mimic the nodular pattern can be also observed in systemic autoimmune diseases, chronic myeloproliferative disorders, toxic myelopathy and viral infections [47]. In contrast to benign lymphoid aggregates in the bone marrow, the nodular aggregates of CLL are less compact and have irregular borders with neoplastic lymphoid cells infiltrating into the surrounding space. Bone marrow can also have proliferation centres or pseudofollicles (Fig. 7.6). In the interstitial pattern the lymphocytes are admixed with the normal hematopoietic elements without much effacement of the architecture. In nodular and interstitial pattern of infiltration normal haematopoiesis is retained. A combination of these patterns is probably the commonest especially a combination of interstitial and nodular pattern [40] (Fig. 7.7). The diffuse infiltration was thought to be associated with a poorer prognosis with a median survival time of 28 months compared to 90 months in nodular and 46 months in interstitial pattern [40]. It has, however, become clear that a diffuse pattern of marrow infiltration is not an independent indicator of poor outcome as it appears to be associated with other poor prognostic features such as unmutated IGHV gene and ZAP-70 expression [48].
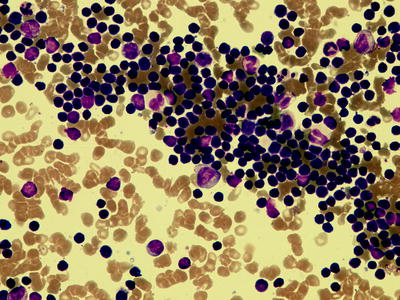
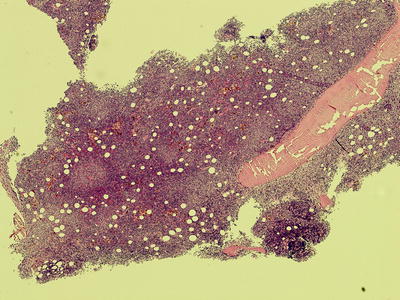
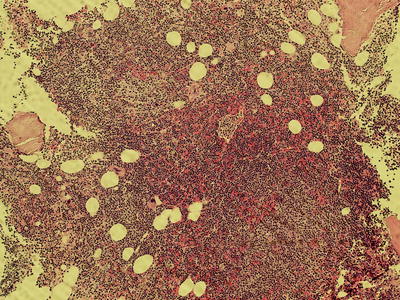
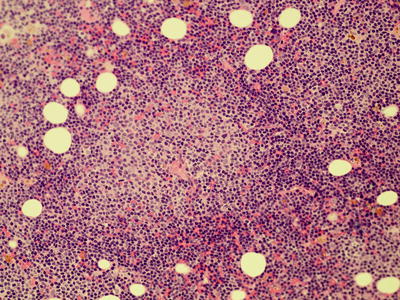
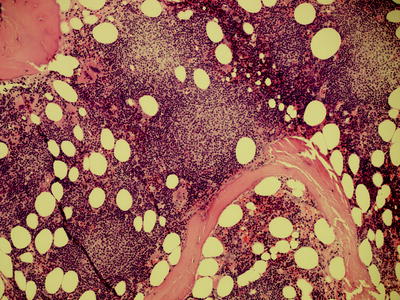
Fig. 7.3
Bone marrow aspirate showing infiltration with CLL cells (500× magnification)
Fig. 7.4
Low power view of trephine biopsy from iliac crest showing diffuse infiltration with CLL cells. The marrow is hypercellular with normal hematopoietic cells and fat spaces replaced by CLL cells. Very prominent proliferation centres (pale areas) are also seen (40× magnification)
Fig. 7.5
High power view of diffuse infiltration of the bone marrow (100× magnification)
Fig. 7.6
Bone marrow trephine biopsy showing a proliferation centre (200× magnification)
Fig. 7.7
Bone marrow trephine biopsy showing extensive interstitial and nodular infiltration with CLL (100× magnification)
Patients can present with symptomatic lymphadenopathy or involved lymph nodes may be found during surgery for other pathologies such as mastectomy for breast cancer, bowel resection or hernia repair. Involved lymph nodes in CLL shows effacement of architecture and can be involved with four growth patterns—pseudofollicular, diffuse, tumour forming and paraimmunoblastic. Three types of cells are seen in these infiltrations: small lymphocytes, prolymphocytes and paraimmunoblasts. The morphology of small lymphocytes and prolymphocytes is similar to that in the peripheral blood. Occasionally the nucleus in small lymphocytes can be cleaved mimicking that of a centrocyte which could be misinterpreted as mantle cell lymphoma. Paraimmunoblasts are large cells, with a wide rim of cytoplasm. The nucleus is round or oval with fine chromatin, prominent central nucleolus and the mitotic figures ranging from low to moderate. In diffuse pattern of growth small lymphocytes predominate over the other population of cells.
Pseudofollicular pattern is the most common pattern which accounts for 85 % [49, 50]. Pseudofollicles or proliferation centres are characteristic features of CLL. At low power these appears as ill-defined paler staining areas (Fig. 7.8) which at high power are seen to comprise a mixture of prolymphocytes and paraimmunoblasts (Fig. 7.9). The CLL cells in the pseudofollicles have a unique phenotype. Significantly they express the proliferation marker Ki-67 along with other markers such as MUM-1, Oct-1, Bob-1 and CD71 [51] (Fig. 7.10). They are also CD5 positive and stain more intensely for CD20 and CD23 than lymphocytes in the surrounding areas [52]. Expression of cyclin D1 has been noted in tumour cells in proliferation centre by using rabbit monoclonal antibodies [53]. Even though the name pseudofollicle is derived from the fact that they mimic germinal centres of a reactive lymph node, their histological appearance and immunochemistry differ considerably. Pseudofollicles are CD10-, Bcl-6-, Bcl-2+ unlike germinal centres, but both are positive for survivin, an anti-apoptotic protein [54]. Survivin is usually expressed by CLL cells in the proliferation centre but not in the peripheral blood. Their expression in the peripheral blood CLL cells predicts for an aggressive behaviour of the disease [55]. The spleen is usually involved as a part of generalized disorder, but very rare cases of isolated splenic involvement are also reported. Clinically patients can have splenomegaly varying from mild to massive. In one report pathological spleens removed weighed up to 4,130 g [56]. Even with massive splenomegaly the occurrence of splenic infarction is very rare in CLL. On gross examination of the cut surface of the spleen there will be uniformly distributed miliary white nodules ranging from 0.2 to 1.5 cm in diameter. In infiltrated spleen, normal white pulp will be completely replaced and the remnants of B-cell follicles are identified only by special staining for follicular dendritic cells. Unlike in reactive changes in CLL there will also be varying involvement of the red pulp area [57]. The peri-arteriolar tumour will extend along the capillaries to the red pulp. In massive involvement the sinuses will be disrupted. Rarely red pulp involvement can be more prominent than the white pulp involvement which is more common in hairy cell leukaemia. Lymphocytic infiltration within dense fibrous trabeculae and subendothelial regions of trabecular veins is found in SLL/CLL and other malignant infiltrations but not in reactive changes of spleen. Proliferation centres are seen in spleen but are less prominent than in lymph nodes. Iron pigment may be prominent because of haemolysis.
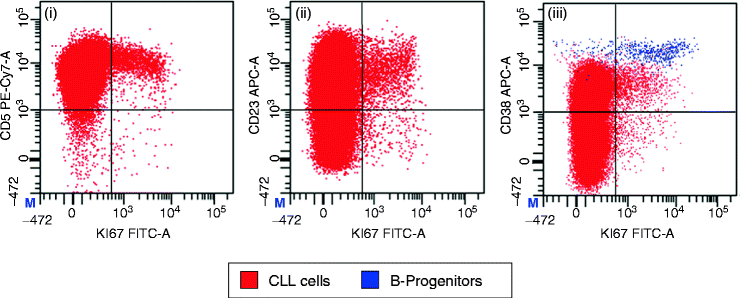
Fig. 7.8
Low power view of the pseudofollicles or proliferation centres in lymph node biopsy which appears as ill defined paler staining areas (magnification 40×)
Fig. 7.9
High power view of the pseudofollicles in lymph nodes (magnification 200×)
Fig. 7.10
Proliferation in CLL. There is no evidence of a discrete proliferative fraction in CLL, but the cells in cycle have restricted expression of several markers. Cycling cells are identified by expression of Ki67. Both CD5 (1) and CD23 (2) expression are typically strong in Ki67+ CLL cells. CD38 (3) expression is also usually strong, but care must be taken in interpreting proliferation in the bone marrow as residual B-progenitors (shown in blue) will have strong expression of both CD38 and Ki67
The liver can be involved in CLL but is less commonly observed than the organs described previously. CLL preferentially infiltrate the portal tracts, but sinusoidal and nodular distributions are also described. CLL infiltration could be confused with lymphocytic infiltration in chronic hepatitis or early stages of primary biliary cirrhosis. But CLL cells are cytologically uniform and immunohistochemistry usually distinguishes them.
Histopathology of Richter’s Transformation
Richter syndrome (RS) is the clinico-pathologic transformation of CLL to an aggressive type of lymphoma, most commonly diffuse large B-cell lymphoma (DLBCL) but rarely transformation into classical Hodgkin’s lymphoma can occur. Incidence of transformation to RS in patients with CLL ranges from 2.2 % to 8 % [58]. Transformation can be clonally related to CLL, which accounts for most of the transformations, or can be unrelated [59–62]. In the latter instances the tumour may be Epstein–Barr virus (EBV)-associated phenomenon driven by both disease and treatment-related immunosupression analogous to post-transplant lymphoproliferative disorders (Fig. 7.11). Clonally unrelated EBV-driven lymphoproliferative disorders have been described in other B-cell disorders like Waldenstrom macroglobulinemia and mantle cell lymphoma [62]. EBV-positive mucocutaneous ulcers with Hodgkin-like histological features have recently been described to be associated with immunosuppressed host [63]. These ulcers are also seen in other lymphoproliferative disorders (unpublished data). These transformations can be generalized or restricted to a single site. Tissue biopsy is essential for a diagnosis of RS. Morphological features of these transformed DLBCL are similar to de novo DLBCL (Figs. 7.12 and 7.13). Almost 80 % of these transformed DLBCL are activated B-cell (ABC) type lymphoma and the rest of them are germinal centre (GC) type. The GC type might imply a de novo unrelated disease [64]. The morphology of this transformed disease should not be confused with increased prolymphocytes in patients with progressive disease. Another possibility where both diseases are not infrequently seen simultaneously are the finding of CLL cells in the bone marrow performed during the staging for DLBCL and usually these DLBCL are clearly unrelated to the CLL population.
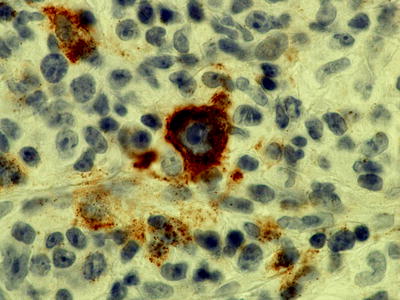
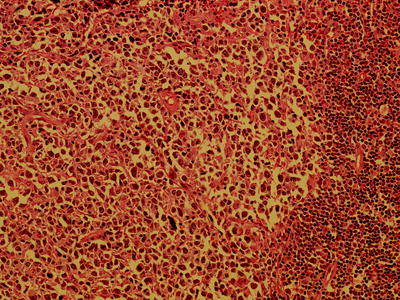
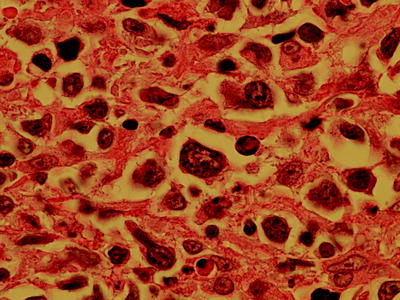
Fig. 7.11
Immunohistochemistry showing latent membrane protein (LMP) staining in large cell transformation of CLL suggesting EBV-driven disease (magnification 1,000×)
Fig. 7.12
High power view of transformation of CLL to diffuse large B-cell lymphoma (magnification 200×)
Fig. 7.13
Transformation of CLL to diffuse large B-cell lymphoma (magnification 1,000×)
Classical Hodgkin’s lymphoma can also occur as an EBV-associated phenomenon or as an unrelated disease (Fig. 7.14). On immunophenotyping they are CD30+, MUM1+ and CD15+/− (Fig. 7.15). B-cell phenotype is characteristically downregulated.
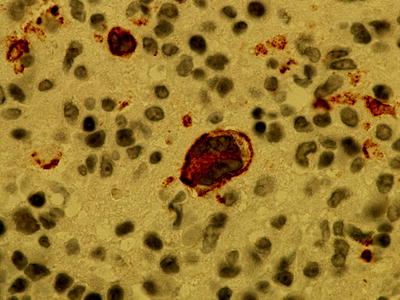
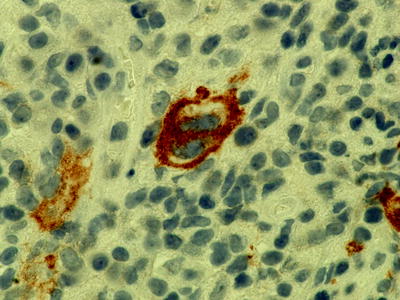
Fig. 7.14
Immunohistochemistry showing latent membrane protein (LMP) staining in Hodgkin’s transformation of CLL suggesting EBV-driven disease (magnification 1,000×)
Fig. 7.15
Immunohistochemistry showing CD30-positive Reed–Sternberg cells in Hodgkin’s transformation of CLL (magnification 1,000×)
Immunophenotyping
Currently immunophenotyping is the most useful diagnostic technique available to evaluate various aspects of CLL. It is helpful in various ways including (1) the differential diagnosis of B- and T-cell LPD and reactive conditions; (2) the identification of therapeutic targets and (3) the development of better prognostic markers as well as (4) to facilitate the assessment of minimal residual disease (MRD) after treatment. Phenotyping of CLL and related disorders can be done either by flow cytometry or immunohistochemistry depending on the tissue sample. Immunophenotyping studies, principally by flow cytometry, were first reported in CLL in the early 1980s [65–67]. These studies identified the co-expression of CD5 (Leu1) and pan-B-cell markers as the phenotypic hallmark of CLL. Subsequent studies have further clarified the immunophenotypic features of CLL. Flow cytometry can be used for diagnosis from most tissues. It should be performed in all cases of unexplained persistent lymphocytosis. From solid tissues such as lymph nodes, a cellular suspension can be used for flow cytometry by disaggregating a sample from fresh specimen. A quick diagnosis can be established by this method. There is a considerable overlap of the diagnostic and prognostic antigens used in both methods.
Diagnostic Markers
The characteristic immunophenotype for CLL includes weak to moderate expression of surface Ig (sIg), which is usually IgD alone or in combination with IgM together with κ or λ light-chain restriction and expression of B-cell-associated antigens (CD19, CD20, CD22 and CD79a) along with CD5, CD23 and CD43. Expression of other Ig heavy chains, IgG or IgA, is infrequent. The intensity of expression is very low and this helps to distinguish CLL from other chronic B-cell disorders. Identification of B-cell clonality is important in order to distinguish neoplastic B-cell disorders from polyclonal B-cell lymphocytosis which is a benign condition where the lymphocytosis can be up to as high as 30 × 109/L and the morphological features could be suggestive of atypical CLL or other neoplastic B-cell disorders.
The expression of the pan-B-cell antigens shows a characteristic pattern in CLL. The immunophenotype of typical CLL is very different from other B-lymphoproliferative disorders and knowledge of the individual antigenic variations compared to other disorders is helpful in explaining some of the features of the disease.
CD19 Expression
CD19 is a pan-B-cell marker which is present on CLL cells. CD19 is an important molecule in B-cell activation. It functions as an adaptor-like protein, mediating the recruitment and activation of signalling molecules to BCR microclusters [68].
CD20 and FMC7 Expression
CD20, a phosphoprotein that may act as calcium channel and plays an important role in cell-cycle progression and differentiation, is dim on CLL cells. CD20 is expressed on all stages of B-cell development except in pro-B-cells or plasma cells. This is a crucial molecule in terms of treatment for CLL as it is the target of the monoclonal antibody rituximab. CD20 is characteristically expressed at low levels in CLL compared to the other B-LPD’s and this feature is useful to help differentiate CLL. FMC7 is another marker, which is negative in CLL, thereby helping to differentiate it from other mature B-cell neoplasms. Even though earlier studies suggested that FMC7 is an independent antigen [69, 70], later it was proved that FMC7 is an epitope on the CD20 molecule [71]. In a diagnostic setting, FMC7 can be useful for distinguishing CLL from other B-cell lymphoproliferative disorders. Considering the fact that FMC7 is an epitope on CD20 molecule, there have been few reports suggesting that it has no discriminatory power over CD20, especially if CD20 antigen density is also taken into consideration [71, 72]. FMC7 can be positive in 15 % of CLL patients, who also have an atypical cell morphology bright CD20 and sIg expression and clinically they may have a more aggressive course compared with FMC7-negative cases.
B-cell Receptor Complex
Early reports suggested that most CLL cases were CD79b negative, but with the use of a phycoerythrin-conjugated CD79b monoclonal antibody has shown that CD79b is expressed weakly in most CLL cases. The level of CD79b, also known as B29, directly correlates with the level of sIg expression in CLL [73]. CD79b in association with CD79a plays a major role in BCR complex formation. This multimeric complex translates specific antigenic stimulation through the surface immunoglobulin into a B-cell response [74]. Low expression of CD79b has been attributed to the development of mutations in the coding sequences of the B29 gene in CLL that produce a truncated form of the protein [75]. This is a very helpful marker in differentiating CLL from other B-cell malignancies as the surface immunoglobulin complex, including CD79b, is strongly expressed in non-CLL LPDs.
CD22 is typically expressed at a low density in CLL. It is a BCR-associated transmembrane protein, the cytoplasmic tail of which contains three immunoreceptor tyrosine-based inhibitory motifs. These motifs are phosphorylated upon BCR-cross linking, and can act as negative regulator of signalling from the BCR. So the under-expression of these two molecules may explain the aberrant signal transduction in CLL cells similar to that of anergic normal B lymphocytes.
CD5 Expression
CD5 is an antigen that is consistently positive in CLL cells and is one of the hallmarks of the disease. CD5 is a pan-T-cell marker. CD5 is also present in a proportion of normal B cells and these are usually found in mantle zones of secondary lymphoid follicles. Up to 30 % of normal peripheral blood B cells express CD5. Regenerating naïve B cells express CD5, such that up to 90 % of normal B cells express CD5 during recovery after high dose therapy [76]. CD5 + B cells have been implicated in producing autoantibodies and it is interesting to note that CLL has a high frequency of autoimmune phenomenon.
CD23 Expression
CD23 is yet another molecule expressed on CLL cells and this is a characteristic immunophenotypic hallmark of B-CLL. CD23 is a 45-kd transmembrane glycoprotein that functions as a low-affinity receptor for IgE and as an adhesion molecule. It is expressed by normal naïve B cells. CD23 is lost during the GC reaction and memory B cells do not express CD23. In CLL it is closely associated with proliferation as Ki67+ CLL cells express the highest levels of CD23 [77]. There are two isoforms of CD23: CD23a which is restricted to B cells and CD23b which could be expressed by B cells as well as other haematopoietic cells including monocytes/macrophages, T cells, eosinophils and platelets when they are stimulated [78]. In vitro data suggest that induction of CD23 will increase intracellular nitric oxide, which protects the CLL cells from apoptosis [79]. High levels of soluble CD23 are found in sera from CLL patients, which have been directly correlated with disease activity [80], [81]. Soluble CD23 serves several functions including extending the survival of B lymphocytes and the induction of differentiation and proliferation of several cell subtypes, including B lymphocytes. Some studies have shown that the expression of CD23 is significantly higher in the prolymphocytoid large cells present in the proliferating centres than in the small lymphocytes, suggesting that the former are the main source of the soluble levels of this molecule detected in the serum [82].
Bcl-2 Expression
Another important molecule over-expressed in CLL is BCL-2, which is a key molecule regulating programmed cell death. It seems likely that the over-expression of Bcl-2 in CLL leads to a resistance to apoptosis and may be critical to the pathophysiology of the disease. In follicular lymphoma Bcl-2 over-expression was linked to t(14;18) where the Bcl-2 open reading frame fuses with Ig regulatory sequences that expose it to the direct influence of Ig promoter, leading to over-expression of Bcl-2. This mechanism cannot explain the Bcl-2 over-expression in CLL as t(14;18) is not a usual finding in CLL [83]. The Bcl-2 over-expression may be related to hypomethylation of DNA in CLL [84].
CD43 Expression
Leukosialin or CD43 is a major sialoglycoprotein on the surface of T cells, myeloid cells, monocytes and small B-cell subset, which appears to be an important molecule for immune function and may be part of a physiologic ligand–receptor complex involved in T-cell activation. It is aberrantly expressed in various B-cell malignancies such as CLL, mantle cell lymphoma, Burkitt’s lymphoma, some marginal zone lymphomas and diffuse large B-cell lymphoma. It is characteristically absent in follicular lymphoma. There are studies suggesting that including CD43 in the diagnosis of CLL will improve diagnostic accuracy [85].
CD52 Expression
CD52 is a glycosylphosphatidylinositol (GPI)-anchored antigen expressed on the B and T lymphocytes, natural killer (NK) cells, monocytes, macrophages and some dendritic cells as well as within the male genital tract. CD52 is an antigen which may not be very important in diagnosis, but the presence of the antigen is crucial in the treatment of CLL especially in relapsed and fludarabine refractory patients as it is the target for the therapeutic monoclonal antibody alemtuzumab.
Therefore the combination of all of these various markers defines CLL and ensures that the differentiation of the disease from other lymphoproliferative disorders is usually relatively straightforward.
Prognostic Markers
CD38 Expression
CD38, a molecule expressed in a variety of haematopoietic cells, including thymic cells, stem cells, activated T cells and B cells and plasma cells, is an important prognostic marker in CLL. CD38 was initially considered as a possible surrogate marker for IvgH mutational status as it is a relatively straightforward marker to analyse routinely. Even though the initial studies suggested that it could be used as a surrogate marker for mutational status, as CD38 positivity was associated with unmutated group and has got a poor prognosis compared to CD38-negative group which is associated with mutated group and better prognosis, more recent reports indicate that CD38 is an independent prognostic marker in CLL and there are other studies suggesting that combination of CD38 and IGHV mutational status had an even greater prognostic power than either marker alone [86–92]. The pattern of CD38 expression can be homogenously positive, homogenously negative or can be bimodal where subsets of CD38-positive and CD38-negative cells seen in the same population [93]. The prognostic significance of this bimodel pattern is not very clear.
Zeta-associated Protein 70
ZAP-70 is another molecule which is associated with the lack of somatic mutations of the immunoglobulin gene and therefore a poor prognosis. However, the recent interest in ZAP-70 is that it may well play a role in the intra-cellular signalling through the BCR and therefore appears to be important in the pathophysiology of the disease. ZAP-70 is a member of the SYK-ZAP-70 protein tyrosine kinase family which is normally expressed in T cells and NK cells and has a critical role in the initiation of T-cell signalling. Gene expression profile studies have found it to be over-expressed in CLL cells with unmutated IGHV genes [94]. Some studies have shown a concordance between 72 % and 95 % when it is used as a surrogate marker [95–97]. But another study has shown that analysing CD38, ZAP70 and IvgH mutational status together will give a more discriminatory prediction of time to first treatment and overall survival. Recently agents have been developed which interfere with the intra-cellular signalling in CLL and these agents appear to have biological activity.
P53 Dysfunction and Chemotherapy Resistance
Another important prognostic marker that can be detected by immunohistochemistry, immunocytochemistry or flow cytometry is P53 protein expression. The p53 gene is located on short arm of chromosome 17 at band 13, and plays an integral role inducing apoptosis or cell-cycle arrest in cells whose DNA is damaged by either chemotherapy or radiation. In CLL functional dysregulation of p53 can occur directly due to deletion or mutation on the 17p chromosome or indirectly due to defects in the regulatory genes, e.g., ATM gene located in 11q region. The wild-type p53 protein is normally undetectable by immunohistochemical analysis using anti-p53 monoclonal antibodies; however, the mutated p53 is detected by immunohistochemical methods as this protein has a prolonged half-life. This may be due to the fact that wild-type p53 protein is targeted to MDM-2-mediated ubiquitination and subsequent degradation [98]. This abnormal p53 protein is expressed in both mutation at the chromosomal region and hemizygous deletion of the region [99, 100]. At genetic level p53 abnormalities are detected by FISH or direct gene sequencing and at protein level by immunohistochemistry, immunocytochemistry or flow cytometry [101–103]. It appears that all these methods have the same significance in assessing the prognostic significance in CLL. 17p chromosomal anomalies are associated with poor prognosis and chemotherapy resistance in CLL and the incidence of p53 abnormalities is much more frequent in chemotherapy resistant CLL compared with de novo cases.
From the diagnostic perspective a single marker could not differentiate CLL from normal cells and other B-cell lymphoproliferative disorders. But by combining a set of markers it is possible to distinguish CLL from other B-cell disorders evolving with a leukaemic picture. A scoring system has been widely used since it was first reported in 1994 which includes CD5, CD23 and FMC7 expression, as well as the level of intensity of sIg and CD22 or CD79b [104]. A score of 1 is given if CD5 and CD23 are positive, FMC is negative, SIg is weakly positive and CD22 or CD79b is weakly positive or negative. The scores for CLL range from 3 to 5 while in the other B-cell disorders they range from 0 to 2. However, this system of scoring has become less relevant with the improved number of markers, molecular techniques and a greater understanding of the disease pathophysiology. In current practice a combination of techniques are used to come to a final diagnosis in difficult cases, for example FISH analysis to detect t(11;14) which positively identifies mantle cell lymphoma in cases which are CD5 positive but CD23 negative or weak. Therefore such Scoring systems for the diagnosis of CLL are not flexible enough and are probably no longer necessary.
MRD by Flow Cytometry
There is convincing evidence that eradication of MRD is associated with an improved outcome in treatment of CLL. MRD can be assessed by multi-colour flow cytometry or real-time quantitative allele-specific oligonucleotide Immunoglobulin heavy chain gene polymerase chain reaction (RQ-ASO IgH-PCR). The initial methods of evaluation of MRD by flow cytometry were not as sensitive as RQ-ASO IgH-PCR which can detect CLL cells down to 0.001 %. But RQ-ASO IgH-PCR as it is expensive, labour-intensive, and cannot be performed unless pre-treatment material is available. In 2007, the European Research Initiative on CLL (ERIC) proposed an international standardized approach after analysing various combinations of antibodies and comparing them against the ASO-PCR technique [105]. After analysing 728 paired blood and bone marrow samples, they derived several conclusions: (1) Blood analysis was equally or more sensitive than marrow in 92 % of samples, but marrow analysis was necessary to detect MRD within 3 months of antibody therapy; (2) The κ/λ/CD19/CD5 combination can be used to screen samples and avoid extended analysis in cases with clear evidence of residual disease where all B cells are CD5+ with light-chain restriction; (3) A CD45/CD14/CD19/CD3 combination or an equivalent can be used to provide a control for CLL cell enumeration and to define the limit of detection; (4) The combination of CD5/CD19 with CD20/CD38, CD81/CD22 and CD79b/CD43 is the best panel to detect MRD with low inter-laboratory variation, low false detection rates, and an accuracy of 95.7 % (Fig. 7.16).
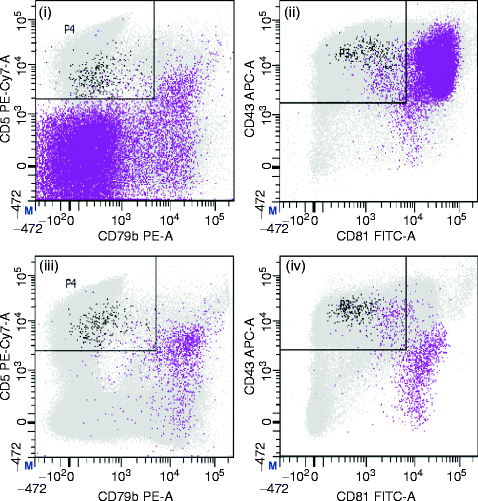
Fig. 7.16
MRD analysis in CLL. The analysis of residual disease is greatly facilitated by multi-parameter flow cytometry. The plots show a case with a very low level of residual disease after treatment. Normal leucocytes are shown in grey, normal B-cells in purple and CLL cells in black. The CLL cells represent 0.06 % of total leucocytes in both peripheral blood and bone marrow whereas total B-cells represent 0.4 % and 5.4 % of leucocytes in blood and marrow, respectively. In the bone marrow—(1) and (2)—the vast majority of normal B cells are progenitors with very strong expression of CD43 and CD81 but no CD5 and weak CD79b expression. In the peripheral blood—plots (3) and (4)—the vast majority of normal B cells are mature naive B cells with weak expression of CD43, moderate expression of CD81 and strong expression of CD79b. Immediately after treatment, a large proportion of normal regenerative naive B cells co-express CD5
Differential Diagnosis (Table 7.1)
Table 7.1
Differential diagnosis of CLL-immunophenotypic features
CLL | Mantle | MZL/LPL/WM/SLVL | PLL | HCL | FL | |
|---|---|---|---|---|---|---|
SIg | Weak | Strong | Strong | Strong | Strong | Positive |
CD5 | Positive | Positive | Negative | Negative or positive | Negative | Negative |
CD23 | Positive | Negative or weak | Usually negative | Positive | Negative or weak | Variable |
FMC7 | Negative or weak | Positive | Positive | Positive | Strong | Positive |
CD79b | Negative or weak | Positive | Positive | Positive | Positive | Positive |
CD20 | Weak | Positive | Positive | Strong | Strong | Positive |
CD22 | Negative or weak | Positive | Positive | Strong | Strong | Positive |
CD19 | Positive | Positive | Positive | Strong | Strong | Weak |
CD79a | Positive | Positive | Positive | Strong | Positive | Positive |
CD43 | Positive | Positive | Negative | Positive | Variable | Negative |
Cd11c | Variable | Weak or negative | Variable | Weak | Positive | Negative |
CD10 | Negative | Negative | Negative | Negative | Variable | Positive |
CD103 | Negative | Negative | Negative | Negative | Positive | Negative |
CD25 | Variable | Variable | Variable | Negative or weak | Positive | Negative |
Cyclin D1 | Negative | Positive | Negative | Negative | Weak | Negative |
SOX11 | Negative | Positive | Negative | Negative | Weak | Negative |
Differential diagnosis for CLL varies depending on the clinical scenarios. For presentation as lymphocytosis and cytopenias the main diseases to be differentiated are mantle cell lymphoma, marginal zone lymphoma and prolymphocytic leukaemia (PLL) (Fig. 7.17). Very rarely splenic diffuse red pulp small B-cell lymphoma, leukaemic presentation of follicular lymphoma and hairy cell leukaemia can give diagnostic confusions. Immunophenotyping using flow cytometry would be the main method for differentiating these disorders. When CLL presents as predominant lymphadenopathy, follicular lymphoma and diffuse large cell lymphoma need to be clearly differentiated. In most cases morphological features and immunohistochemistry are adequate to differentiate these disorders. Lymphocytic infiltration at extranodal sites can be a feature of any of the previously mentioned conditions and again morphology and immunohistochemistry serve as a differentiating tool in most cases.
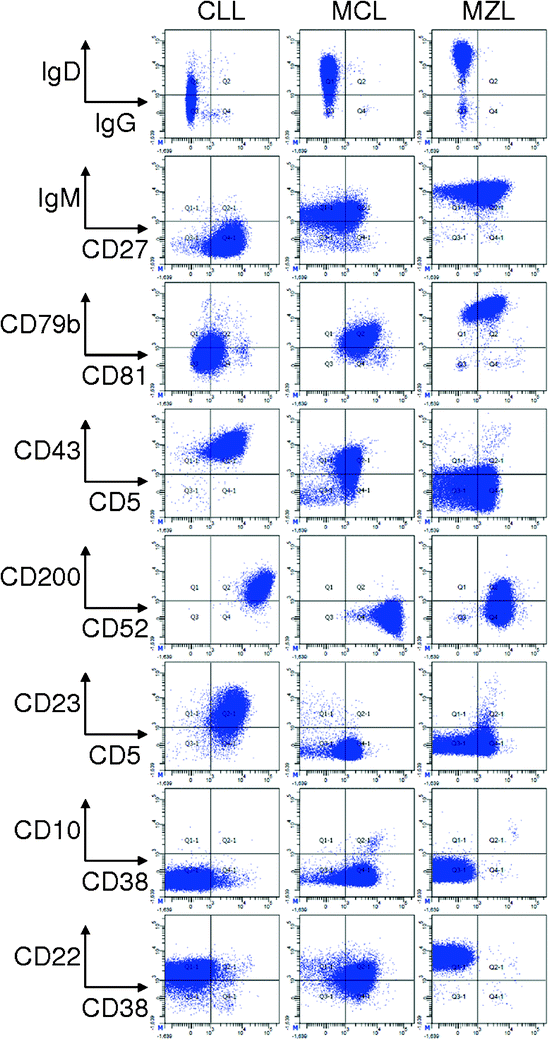
Fig. 7.17




Representative plots from a case of CLL (left column), Mantle Cell Lymphoma (middle column) and a marginal zone lymphoma with CD5 expression (right column)
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
