Like most biologic processes, the normal patterns of silencing can be altered, resulting in the development of disease states. Thus, activation of genes normally not expressed, or silencing of a gene that should be expressed, can contribute to the dysregulation of gene function that characterizes cancer and, when stably present, represent epigenetic alterations.4–7 Most studies have focused on the silencing of normally expressed genes. For the purposes of understanding the rationale behind epigenetic therapy, it is important to understand the mechanisms through which such silencing occurs. Alterations in gene expression associated with epigenetic changes that give rise to a growth advantage would be expected to be selected for in the host tissue, leading to progressive dysregulated growth of the tumor. Such dysregulation is commonly associated with increases in promoter region DNA methylation and is associated with repressive chromatin changes.
Changes in DNA Methylation
The importance of abnormal cytosine methylation and gene silencing has been clearly established in the past 2 decades and been shown convincingly to be involved in cancer development.4–7 The CpG dinucleotide, usually underrepresented in the genome, is clustered in the promoter regions of approximately 50% of human genes in regions termed CpG islands. These regions are largely protected from DNA methylation in normal cells, with the exception of genes on the inactive X chromosome and imprinted genes.16 This protection is critical, because the methylation of promoter region CpG islands is associated with a loss of gene expression.4–7 Abnormal de novo DNA methylation of gene promoter CpG islands is a very frequent abnormality in virtually all cancer types and is associated with a process that can serve as an alternative mechanism for loss of tumor suppressor gene function.4–7 Although a limited number of classic tumor suppressor genes can be affected by this process, a patient’s individual cancer may harbor hundreds of such genes.4–7 Which of these latter genes are drivers of cancer, individually or in groups, versus those which are passengers reflecting only the widespread effects of a global epigenetic abnormality is a leading question in the field and the target of much research.5,6 A clue to the importance of at least groups of the previous DNA hypermethylated genes may come from the fact that an inordinate number of them are involved in holding normal embryonic and adult stem cells in the self-renewal state and/or rendering such cells refractory to differentiation cues.17,18 Normally, these genes are then in a poised expression state and can be induced to be activated or repressed as needed for changes in cell state.18 Abnormal promoter DNA methylation of such genes renders them more repressed and could be a factor in the fact that cancers inevitably exhibit cell populations with enhanced self-renewal or refractoriness to full differentiation.18
Recent studies have also suggested that DNA regions other than promoter CpG islands may undergo changes of DNA methylation in cancer. For example, non–CpG-rich sequences surrounding promoter CpG islands, termed CpG island shores, are abnormally methylated in cancers19 and may be altered in stem cell populations.20 Thus, the relative cancer specificity of changes of DNA methylation in multiple CpG regions makes reversal of these changes by targeting DNA methyltransferases, the enzymes that catalyze DNA methylation, logical for cancer therapeutics.
As a key example of the previous points, perhaps the most studied tumor suppressor gene for promoter hypermethylation is the p16 gene, currently designated CDKN2A, a cyclin-dependent kinase inhibitor that functions in the regulation of the phosphorylation of the Rb protein. Hypermethylation associated with loss of expression of the CDKN2A gene has been found to be one of the most frequent alterations in neoplasia being common in the lung, head and neck, gliomas, colorectal, and breast carcinomas21,22 and other cancer types. A member of the same gene family, p15 or CDKN2B, also regulates Rb and is silenced in association with promoter methylation in many forms of leukemia and in the chronic myeloid neoplasm myelodysplastic syndrome (MDS).23 These two previous changes are of much relevance for the clinical uses of epigenetic therapies discussed later.
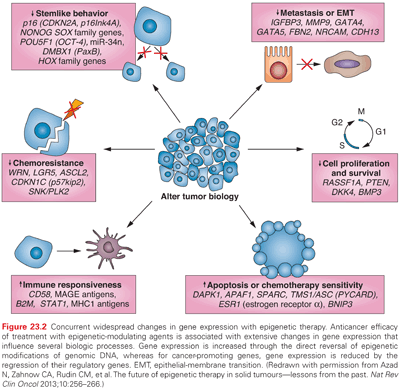
As mentioned, many hundreds of genes may be inactivated in a single cancer by promoter methylation,5,6,18,24 providing potential targets for gene reactivation using epigenetic therapies.25–27 The latter represents one of the potential ways in which epigenetic therapy may be effective: Multiple genes and gene pathways, all repressed by changes in DNA methylation and chromatin modification, can be reactivated by DNA methyltransferase inhibitors and histone deacetylase (HDAC) inhibitors (HDACi), thereby restoring normal cell cycle control, differentiation, and apoptotic signaling (Fig. 23.2).8,26,28 In general, methylated CpG islands are not capable of the initiation of transcription unless the methylation signal can be overridden by alterations in factors that modulate chromatin, such as the removal of methylated cytosine-binding proteins. However, reversal of DNA methylation with secondary changes in histone modification or directed reversal of repressive histone modifications represent a target for epigenetic therapies.8,26,28
Most studies of DNA methylation, particularly in the study of cancer, have focused on CpG island promoter methylation. However, about 40% of human genes do not contain bona fide CpG islands in their promoters.29 The primary focus on CpG islands has resulted from the clear demonstration that CpG-island promoter methylation permanently silences genes both physiologically and pathologically in mammalian cells. However, recent work has shown correlations between tissue-specific expression and methylation of non-CpG islands, including, for example, the maspin gene,30 and as mentioned previously, regions near CpG islands,19,20 suggesting that many additional genes could be regulated, either normally or abnormally, by changes in DNA methylation.
An exciting new area of DNA methylation research involves the role of this change in regulating gene enhancers: small DNA regions that regulate the expression of multiple target genes.31–33 The presence of DNA methylation in these areas, which can reside considerable distances from the genes that are being regulated, generally works together with histone modifications to mediate a repressive state for that enhancer.31–33 The status of enhancers is also emerging as important for cancer risk states.34
Chromatin in Gene Regulation
Heritable gene silencing involves the interplay between DNA methylation and histone covalent modifications. Complexes of proteins that can regulate how nucleosomes are positioned perform nucleosomal remodeling.35–37 What was initially termed the histone code, with reference to how histones are modified, has emerged to be much more complex than originally envisioned. An explosion of research findings during the last several years now allows for an appreciation of how the epigenome is controlled by a complex interplay between a myriad of posttranslational histone modifications that occur on key amino acid residues of these proteins.37 Acetylation, deacetylation, methylation, phosphorylation, and other modifications all modify chromatin structure and thereby alter gene expression.38 Some of the enzymes that catalyze these modifications include HDACs, histone methyltransferases (HMT), and most recently, histone demethylases.13,14,39,40 These modifications help establish heritable states at the start site of genes, but also at enhancers and other transcribed DNA regions not encoding for canonical genes. The latter areas contain noncoding RNAs (ncRNAs) and micro-RNAs (miRNAs), which play key modulatory roles for overall gene expression and protein patterns that can be altered in cancer.41–43 Again, much research is being focused on epigenetic changes in these DNA regions, which may be important to cancer development and, potentially, to cancer management.
A link between covalent histone modifications and DNA methylation has been clearly established.44–46 In this interaction, cytosine methylation attracts methylated DNA-binding proteins and HDACs to methylated CpG sites during chromatin compaction and gene silencing.46,47 In addition, the DNA methylation binding protein (MBD2) interacts with the nucleosomal remodeling complex (NuRD) and directs the complex to methylated DNA.48 This complex also binds HDACs and has recently been identified as a central player for the abnormal silencing of genes associated with promoter DNA hypermethylation in cancer.47 Thus, the three processes of DNA cytosine methylation, histone modification, and nucleosomal remodeling are intimately linked, and alterations in these processes can result in abnormalities of gene expression in cancer-relevant genes.
Enzymes Regulating DNA Methylation and Histone Acetylation
DNA methylation involves the covalent addition of a methyl group to the 5′ position of cytosine. In mammals, three enzymes have been shown to catalyze this transfer of a methyl group from the methyl donor S-adenosylmethionine. Most of the methyltransferase activity present in differentiated cells is derived from the expression of DNMT1.49 This enzyme is thought to be most important in maintaining DNA methylation patterns following DNA replication and thus is referred to as a maintenance methyltransferase. However, the enzyme does possess the ability to methylate previously unmethylated DNA sequences (de novo activity).50 In contrast, the other enzymes, DNMT3a and DNMT3b, are efficient at methylating previously unmethylated DNA and thus are considered de novo methyltransferases. Each of these enzymes possesses a similar catalytic site,51 a fact important for the inhibition of DNMT enzymes by nucleoside analogs, discussed later in this chapter.
DNA methylation is closely associated with changes in the histone modifications. As previously discussed, histone proteins are the central components of the nucleosome, and modifications of the histone tails of core histones are associated with active or repressed chromatin.52 Although it is beyond the scope of this chapter to fully discuss the complex series of modifications to the histone tails of histone H3 and H4, a few well-characterized modifications should be mentioned that are relevant to therapies designed to target epigenetic abnormalities in cancer. In reference to currently investigated epigenetic therapies, changes in histone acetylation are of importance. Acetylation of histones H3 and H4 at key amino acids is associated with the active chromatin present at the promoters of transcribed genes, whereas the absence of histone acetylation is associated with repressed, silenced genes.13,14,53 Histone acetyltransferases (HAT) HDACs have opposing functions to maintain the proper level of histone acetylation for gene expression.13,14,53 HDACs specifically deacetylate the lysine residues of the histone tails, and this deacetylation is associated with condensation of nucleosome positions in what is termed a closed chromatin formation. This scenario is key to transcriptional repression. There are four classes of HDACs.53 Class I HDACs are characterized by their similarity to the yeast Rpd3 HDAC. In humans, this class of enzymes includes HDAC1,-2, -3, and -8. These HDACs are thought to be ubiquitously expressed in tissue throughout the body. In contrast, class II HDACs are similar to yeast Hda1 and include HDAC4, -5, -6,-7, -9, and -10, and they have a greater degree of tissue specificity. Class III HDACs are similar to yeast Sir2 and are set apart from the other classes by their dependence on nicotinamide adenine dinucleotide (NAD+) as a cofactor. Finally, class IV includes HDAC11.53
Of the previously listed HDACs, class I and 2 HDACs have been most closely tied to gene silencing associated with abnormal promoter DNA hypermethylation.48 These are bound to the nucleosome remodeling complex, NuRD.48,49 Experimental decreases in NURD, after use of a DNA demethylating agent, can augment reactivation of many abnormally silenced and DNA hypermethylated genes in colon cancer cells.48 Manipulation of these HDACs is under study in clinical trials, with and without the use of DNA methyltransferase inhibitors, and is discussed later. Another HDAC, SIRT1 in the class III of these proteins, is also involved with gene silencing.54,55 This deacetylase has been linked to silencing of DNA hypermethylated genes, and blocking its activity can be associated with reactivation of such genes.55
Reversal of Layers of Gene Silencing
The interaction between DNA methylation and HDAC activity and repressive chromatin marks in maintaining aberrant silencing of hypermethylated genes in cancer has therapeutic implications for epigenetic therapies. Experimental evidence suggests that DNA methylation functions as a dominant event that stably establishes transcriptional repression. Inhibition of HDAC activity alone, by potent and specific HDACis, does not generally result in the reactivation of aberrantly silenced and densely hypermethylated genes in tumor cells.56 In contrast, treatment with HDACis can reactivate densely silenced genes if the cells are first treated with demethylating drugs, such as 5-azacitidine.56 The clinical implications of this observation are discussed in more detail in the following section (Table 23.1).
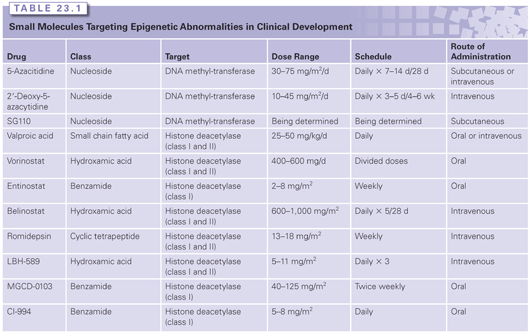
DNA Methyltransferase Inhibitors
Originally synthesized as cytotoxic antimetabolite drugs in the 1960s,57 azacytosine nucleosides were recognized as inhibitors of DNA methylation in the early 1980s. The inhibitors 5-azacitidine (5AC) and 2′-deoxy-5-azacytidine induced muscle, fat, and chondrocyte differentiation in mouse embryo cells, in association with a reversal of DNA methylation.58,59 The incorporation of azacytosine nucleosides into DNA in lieu of cytosine residues was shown to be associated with inhibition of DNMT activity.59,60 DNMT inhibition requires the incorporation of decitabine triphosphate into DNA. The incorporated azacytosine nucleoside forms an irreversible inactive adduct with DNMT. The sequential reversal of DNA methylation then results when DNA replication proceeds in the absence of active DNMT.61 The inhibitor 5AC must be phosphorylated and converted to decitabine diphosphate by ribonucleotide reductase before it can be activated through triphosphorylation, whereas decitabine does not require ribonucleotide reductase. The inhibitor 5AC can also be incorporated into RNA. DNMT2, a misnamed protein that is actually an RNA-specific methyltransferase,62 becomes inhibited, leading to the depletion of methylated tRNA.60 This may contribute to the inhibition of protein synthesis and is a potential difference between azacitidine and decitabine.63 The previous DNA methyltransferase inhibitors not only block the catalytic activities of DNMTs, but also trigger degradation of these proteins, especially DNMTs 1 and 3B.64–68 This latter activity is potentially important for their activities for gene reexpression because each of these two proteins, experimentally, possess transcriptional repression properties independent of their DNA methylation catalytic sites.69,70
The azacytosine nucleosides exhibit complex dose–response characteristics. At low concentrations (0.2 to 1 μM), the epigenetic activities of these drugs predominate, with dose-dependent reversal of DNA methylation71,72 and induction of terminal differentiation in some systems.28,71 As concentrations are increased, DNA damage and apoptosis become more prominent.28,72 Cell lines with 30-fold resistance to the cytotoxic effects of doxifluridine, adriamycin, cyclophosphamide (DAC) continue to reverse methylation in response to this nucleoside, suggesting that the methylation reversing and cytotoxic activities of this compound can be separated.73 The ability of these drugs to inhibit the cell cycle, at least in part through induction of p21WAF1/CIP1 expression, complicates the goal of reversing DNA methylation, because the latter requires DNA replication with the azacytosine nucleoside incorporated into the DNA.
The importance of low doses of the two azacytosine nucleosides to achieve a targeted therapeutic effect has been recently explored in a series of laboratory observations. Transient exposure of both leukemia and solid tumor cells to submicromolar doses induce such cells to undergo cellular reprogramming, accompanied by decreases in ability to clone in long-term self-renewal assays and to grow as explants in immune-incompetent mice.28 These effects occur with partial genome-wide DNA demethylation and changes in gene expression in multiple pathways potentially key for driving tumorigenesis.
The pharmacokinetic properties of the two azacytosine nucleosides are also very important to consider for their clinical use. In this regard, a major potential challenge for their usage is the fact that these drugs are highly unstable in an aqueous solution, resulting in their rapid hydrolysis and resultant inactivation.74 In clinical practice, the drugs must be administered shortly after reconstitution. The drugs are also metabolized by cytidine deaminase,74 leading to a short half-life in plasma. When injected subcutaneously, 5AC reaches a maximal plasma concentration at 30 minutes, with a terminal half-life of 1.5 to 2.3 hours.75,76 At the U.S. Food and Drug Administration (FDA) approved dose of 5AC (75 mg/m2 administered subcutaneously daily for 7 days), peak plasma concentrations were 3 to 5 μM, which is well within the range of DNMT inhibitory concentrations.75,76 Intravenous (IV) administration of the same dose has led to higher peak plasma concentrations (11 μM) with a shorter half-life (approximately 22 minutes).75 DAC given over 1 hour IV at 15 to 20 mg/m2 produced plasma concentrations of 1.1 to 1.6 μM during the infusion,77 whereas in a phase 1 study in patients with thoracic malignancies, patients were treated with escalating doses of decitabine for 72-hour IV infusions for two 35-day cycles. The maximum tolerated total dose was 60 to 75 mg/m2 with neutropenia as the dose-limiting toxicity. Steady-state plasma concentrations ranged from 25 to 40 nM, which is less than those usually used to induce expression of methylated genes in tissue culture models.78 An oral formulation of 5AC has also been studied. The oral bioavailability of oral azacitidine ranged from 6% to 20%. Nonetheless, MDS and acute myelogenous leukemia (AML) patients receiving oral azacitidine developed clinical responses similar to patients receiving parenteral azacitidine. Oral azacitidine has also been safely administered on 14-daily and 21-daily schedules repeated monthly. The extended administration of lower daily doses may provide favorable pharmacodynamics of DNA methylation reversal given the need for ongoing cell cycling to effect methylation reversal.79
SGI-110 is a dinucleoside that acts as a prodrug for decitabine. This drug is being studied in myelodysplasia and AML.80
HISTONE DEACETYLASE INHIBITORS
The increasing recognition of the critical importance of histone modifications in regulating the transcriptional permissively of chromatin has led to intense interest in compounds that can inhibit the activity of HDAC proteins, facilitating the acetylation of lysines associated with transcriptional activation of genes. As with the DNMT inhibitors discussed previously, there are multiple, sometimes dose-dependent, effects of HDACis in preclinical studies. Some of these may truly be epigenetic, others strictly cytotoxic, and others a combination of both.9,81–84 Some actions of HDACis may relate to altering how chromatin is central to the repair of DNA. Thus, at especially high doses, these compounds can blunt efficient repair and even induce DNA breaks.84,85 These effects may underlie cell cycle arrest and induction of cell death as is often observed in preclinical studies of HDACis.81–84
Perhaps novel uses of these drugs may be inferred by results from recent studies suggesting they could be extremely powerful epigenetic therapy agents when used in proper doses, for targeted purposes, and at key time intervals. Recent studies by Settleman and colleagues86 suggest that histone acetylation changes, and thus epigenetic mechanisms, could be a key factor for cancer therapy resistance to both targeted therapy agents and conventional chemotherapy. The mechanisms involved may involve the emergence of drug-tolerant stem-like cells.86 In such cells, gene expression studies suggest that a protein upregulated in resistance is a histone demethylase, which diminishes a key histone modification for active transcription, H3K4methyl.86 A very similar enzyme has been shown in other studies to be central to self-renewal of stem-like melanoma cells.87 Key to the therapies under discussion is that, in the previous drug-resistance studies, low doses of HDACis, could reversibly reduce drug-resistant cells induced by the various anticancer drugs.86 It is essential going forward to sort out which of these effects are dose-related off-target effects and which are desired on-target effects that can be optimized for efficacious therapy strategies.
Types of Histone Deacetylase Inhibitors
Small Chain Fatty Acids
The earliest report of the use of an HDACi to treat leukemia described the treatment of a child with refractory AML with intravenous sodium butyrate, with a concomitant clearance of peripheral blood blast cells and a decrement in bone marrow blasts.88 No responses developed in a subsequent study of nine AML patients who were treated with intravenous butyrate.89 Phase 1 studies of sodium phenylbutyrate (NaPB) in MDS and AML explored 7-day continuous infusions administered monthly or biweekly, and 21-day continuous infusions administered monthly.90,91 At the maximum tolerated dose (375 mg per kilogram per day), the mean steady-state plasma concentration was 0.3 mM, within the range of HDAC inhibition.90–92 Isolated patients developed hematologic improvement in response to NaPB.
Similar to NaPB, valproic acid (VPA) requires near millimolar concentrations to effectively inhibit HDACs. Of 18 patients with MDS or AML with trilineage dysplasia treated with VPA to target plasma concentrations of 0.3 to 0.7 mM, 6 patients developed hematologic improvement.93 Of 20 elderly patients with AML treated with VPA, only 11 could remain in control long enough to be considered evaluable for response. Five had improvement in platelet counts.94 VPA induced hematologic improvement in combination with all-transretinoic acid in two of eight patients treated with AML; a fluorescence in situ hybridization analysis showed definitive evidence of terminal differentiation of the malignant cells.95 A larger study of this combination induced hematologic response in only 2 of 26 elderly patients with AML.96 It appears unlikely that the small chain fatty acids will develop an important role in the treatment of malignancy given the availability of HDACis with vastly greater potency.
Hydroxamic Acids
The FDA approved vorinostat as the first commercially available HDACi. The approval was based on activity of this agent in cutaneous T-cell lymphoma (CTCL). Thirty-three patients with a median number of five prior systemic therapy regimens received one of three dose schedules of vorinostat in a single institution study.97 Eight patients achieved a partial response, with a median time to response of 12 weeks and a median duration of response of 15 weeks. Overall, 45% of patients had relief of pruritus. Fatigue, diarrhea, nausea, and thrombocytopenia were common toxicities. In a multicenter phase 2 trial, 74 patients with relapsed or refractory CTCL were treated with 400 mg daily.98 Similar to the prior study, 29% of patients responded, consisting almost entirely of partial responses. Median time to response was 56 days, and median duration of response was greater than 6 months. In phase 1 trials, responses to vorinostat have developed in other non-Hodgkin’s and Hodgkin’s lymphoma cases.99 More recently, in a trial combining vorinostat with carboplatin and paclitaxel in patients with untreated, advanced, non–small-cell lung cancer (NSCLC), response rates increased significantly from 12.5% to 34%, and a trend to improved progression-free survival and overall survival was observed.100
Panobinostat (LBH589), a cinnamic hydroxamic acid HDACi, reduced peripheral blood blast percentage but did not induce remissions in a phase 1 trial of daily times 7 oral dosing in patients with a variety of relapsed hematologic malignancies.101 Asymptomatic changes in electrocardiographic T waves developed in 80% of treated patients. Gastrointestinal symptoms and thrombocytopenia were common. Panobinostat has recently been approved by the FDA for the treatment of multiple myeloma.102
Cyclic Tetrapeptides
Romidepsin is FDA approved for the treatment of CTCL103 and peripheral T-cell lymphoma.104,105 Antitumor activity, including tumor lysis syndrome, was demonstrated in a phase 1 study that enrolled patients with chronic lymphocytic leukemia and AML, but no complete or partial remissions were seen.75 The administration of romidepsin induces electrocardiographic changes, including T-wave flattening and ST-T wave depression in greater than half of the posttreatment tracings; however, no changes in serum cardiac troponin levels or left ventricular ejection fraction have been reported.106
Benzamides
Entinostat, formerly known as MS-275, was administered weekly times four to patients with relapsed and refractory AML in a phase 1 study. Infections, unsteady gate, and somnolence were dose-limiting toxicities. No clinical responses developed, although improvements in neutrophil counts were observed.107 Entinostat did not increase the response rate in patients with higher risk MDS and AML with MDS-related changes when combined with azacitidine compared to azacitidine alone.108 Most recently, however, studies NSCLC suggest that entinostat could be a valuable therapeutic agent in solid tumors when used with established therapies. When combined with the epidermal growth factor inhibitor erlotinib, in a randomized phase 2 trial for patients with recurrent advanced NSCLC, entinostat was not efficacious alone but appeared to combine with erlotinib to benefit a group of patients whose tumors contained baseline high E-cadherin levels. Overall survival in these latter patients yielded an increased survival benefit of 9.4 versus 5.4 months.109 Finally, entinostat significantly increased survival when combined with an aromatase inhibitor in a phase 2 trial for patients with breast cancer.110
Pharmacodynamic Properties
The administration of oral vorinostat was associated with a transient increase in acetylation of histone H3 in peripheral blood lymphocytes, which peaked at 2 hours post dosing and reverted to baseline by 8 hours; similar changes were observed in the lymph node of a treated patient with lymphoma.99
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


