Fig. 5.1
Overview of NGS sequencers. Full run throughput in gigabases (billion bases) is plotted against single-end read length for the different sequencing platforms, both on a log scale
5.2.1.1 Ion Torrent Sequencers
In 2013, Thermo Fisher Scientific bought the leader in Sanger-based sequencers Life Technologies. Life Technologies released in 2006 their first NGS sequencer, the SOLiD (Sequencing by Oligonucleotide Ligation and Detection) system and the SOLiD 5500 in 2011. Despite significant optimizations of the 5500 system, a high accuracy due to dual reading, the technology failed to move from mid high-throughput to high-throughput as Illumina successfully did. SOLiD sequencers suffered the comparison with HiSeqs because of laborious hands-on, low reliability and throughput, high cost per megabase, and lack of bioinformatics development. Life Technologies acquired then Ion Torrent Systems Inc. in 2010. Ion Torrent Systems Inc. developed an ion semiconductor sequencing technology, a method of DNA sequencing based on the detection of protons released during DNA polymerization. Development of SOLiD systems became since very limited and currently only Ion Torrent systems are sold and further developed by Thermo Fisher Scientific. Ion Torrent sequencers represented 16 % of NGS sequencers installed in the world in 2013, one fourth of Illumina sequencers. Contrary to Illumina, no fluorescence is measured during sequencing reaction but pH. First, libraries are immobilized on hydrogel beads by emulsion PCR. Briefly a single tube PCR reaction containing million of droplets each constituting a single PCR system is performed. In each droplet, one library DNA fragment and one bead are present. The library fragment hybridizes on the bead through the annealing of library adapter to complementary DNA fragments coated on. The PCR replicates more than thousand times the library molecule on the bead. Once the reaction is over, emulsion is broken and beads containing fragments are specifically recovered. This emulsion PCR-based system needs to be improved since more than one library molecule per emulsified PCR system is present in 20–30 % of droplets, leading to a loss of 20–30 % of throughput. Prepared beads are then loaded on Ion chips that are the size of a microprocessor and which contain several millions of wells; each can receive only one bead due to space limitation. This process has been recently shifted from manual to automatic with the commercialization of the Ion Chef in 2014. The ready-to-sequence chip is then loaded in an Ion Torrent sequencer, either the Ion PGM or the Ion Proton. To sequence the library molecules immobilized on beads, the sequencer flows the 4 unmodified dNTPs, dATP, dTTP, dCTP, and dGTP successively including a wash between every flow. When the flowed dNTP is complementary to the next unpaired nucleotide on the template strand, it is incorporated into the growing complementary strand by the DNA polymerase. A proton is released during this DNA polymerization, and the pH of the well in which the bead stands is modified. Each well of the chip has a corresponding ion-sensitive field-effect transistor that measures ion concentration in solution. The sequencer detects and records the pH modification every time a nucleotide is incorporated in a well. At the end of the run, the recorded signals in every well are transformed into DNA sequence corresponding to library fragments.
The two sequencers actually available, the Ion PGM and the Ion Proton, differ only by their throughput (Fig. 5.1) (www.iontorrent.com). The number of wells present in Ion chips determines the throughput of the sequencer. Three chip formats exist for Ion PGM, 314, 316, and 318 chips that can deliver up to 0.6, 3, and 5.5 million reads of maximum 400 nucleotides in 2–7 h. The Ion Proton has only one chip available, the PI chip, the PII being planned to be released in 2015. PI chip delivers up to 82 million reads of up to 200 nucleotides in 2–4 h. These throughputs are insufficient to sequence large genomes or transcriptomes, but these two benchtop sequencers were designed for the clinical diagnosis market. Their major strengths are the cost of devices ($50,000 for a PGM and $149,000 for a Proton) (www.allseq.com), the rapid sequencing that happens in less than 1 day, a scalable throughput, and low operating prices, in part because of the absence of fluorescence. The main limitations of the system are the absence of high-throughput system and the sequencing of homopolymer regions. Contrary to Illumina sequences for which only one base can be added before signal acquisition, if the same base is repeated on a template strand, then multiple nucleotides are incorporated with the Ion Torrent technology. It leads to the release of a higher rate of protons modifying proportionally the pH. A homopolymer of two consecutive identical bases has a signal twice a single nucleotide (100 % increase) which is easy to quantify, but the difference between signals corresponding to 8 and 9 identical successive nucleotides does not differ enough (theoretical increase of 12.5 %) to avoid miscalling. A lot of work is done by Ion Torrent to improve the accuracy of homopolymer sequencing, notably with the recent release of the Hi-Q chemistry.
5.2.1.2 Roche Sequencers
Roche was acquired in 2007 454 Life Sciences, a company founded by Jonathan Rothberg, the founder of Ion Torrent. The high-throughput sequencing technology developed by 454 Life Sciences is based on pyrosequencing technology and is very similar to Ion Torrent technology, but instead of pH modification, fluorescence is emitted upon nucleotide incorporation during DNA polymerization. Libraries are prepared similarly than with Ion Torrent method, and prepared beads are loaded on a Pico TiterPlate, a fiber-optic chip, one bead per well. A cocktail of enzymes, DNA polymerase, ATP sulfurylase, luciferase, and apyrase is added in every well as well as their substrates, adenosine 5’ phosphosulfate (APS) and luciferin. Similarly to Ion Torrent sequencing devices, the 4 nucleotides are sequentially flowed by the sequencer into the chip, and their incorporation generates a signal recorded by the sequencer. When a nucleotide is incorporated to the growing complementary strand of a library molecule by the DNA polymerase, a pyrophosphate is released in the well. The ATP sulfurylase presents in the well converts this pyrophosphate in ATP in the presence of adenosine 5’ phosphosulfate. Through the action of luciferase, luciferin is converted in oxyluciferin that generates an amount of fluorescence proportional to the amount of ATP that corresponds to the number of nucleotides incorporated. Unincorporated nucleotides as well as ATP are then degraded by the apyrase, and another nucleotide is flowed into the chip. At the end of the run, nucleotide sequence of library molecules present in every well is reconstituted. The first 454 sequencer released in 2005 was the Genome Sequencer FLX, and a newer version was released in 2008, the GS FLX + Titanium system (Fig. 5.1) (www.454.com). The last system version generates up to one million reads of up to 1000 bases in 23-h runs for a throughput of 700 megabases (www.allseq.com). This throughput is significantly lower than other sequencers on the market, but the long reads produced make this platform extremely useful for niche applications such as the assembly of de novo sequenced genomes. Later on a less powerful version of the Genome Sequencer FLX system, the GS Junior was released. This device can sequence up to 100,000 reads of up to 700 bases in 10 h and is mainly dedicated to researchers with modest sequencing needs. Due to a high cost per base, a high error rate in homopolymers, and low throughput compared to Illumina and Ion Torrent sequencers, Roche announced in 2013 the shutting down of their sequencing business.
5.2.1.3 The Third Generation of Sequencers
The actual second generation of sequencers suffers from bias and limitations mainly due to the requirement of amplification of library molecules before sequencing. Indeed, the signal (fluorescence, pH) detected by the sensor systems of sequencers needs to be intense to be detected. Since nucleotide incorporation does not happen correctly in all molecules of the same cluster or beads, dephasing of sequencing signal occurs along with the growing of sequenced strand and prevents the sequencing of accurate longer reads. Short reads produced by actual sequencers are not sufficient to generate long continuous assemblies of complex genomes that contain numerous repetitive sequences (transposable elements, high copy genes, centromeric/telomeric sequences, segmental duplications). The third-generation sequencers aim to be able to sequence single molecules allowing direct sequencing of nucleic acids, long reads, no bias due to amplification (GC content), and absolute quantification. Currently, only one third-generation sequencer has been released yet, the PacBio RS by Pacific Biosciences in 2010 and, its latest version, the PacBio RS II in 2013 (Fig. 5.1) (www.pacificbiosciences.com). It can generate reads of up to 15,000 bases in real time but with a reduced throughput of 50,000 reads (up to 1 gigabase sequenced) in up to 240-min run and with a much lower quality compared to second-generation sequencers. Latest version of reagents, protocol of library preparation, and system produce reads with an average length >10 kilobases (www.allseq.com). The optical system that records the sequencing signal is essentially taking a movie of fluorescent nucleotide incorporation. Briefly, single molecule is bound to a single DNA polymerase coated in a zero-mode waveguide (ZMW) on a sequencing small plastic cell called single-molecule real-time cell. ZMW is a structure that captures signal only from nucleotides that are being incorporated, while signal emitted by unincorporated is filtered out. The main applications of this system are for applications that required long reads such as de novo sequencing of small genomes. The rate of nucleotide incorporation is 2–3 bases per second, and the measure of nucleotide incorporation rate allows the determination of modification status of the template nucleotide (5-mC, 5-hmC, etc.), making this sequencer interesting for epigenetic studies. Advantages are low cost of run and single-molecule sequencing, but the main weaknesses are a high machine cost, a low throughput, and low raw accuracy of reads even if contrary to second generation of sequencers; sequencing errors are stochastic and the use of multiple reads gives high accurate consensus reads.
One of the most promising types of third-generation sequencers is based on nanopores. Several companies such as Illumina and Roche are developing or have interest in nanopore-based sequencers. Actually, the most advanced project is conducted by Oxford Nanopore Technologies, a UK-based company that has worked on nanopores for almost 20 years. In 2013, they selected genomic centers to evaluate the technology of their first nanopore-based sequencer, the MinION, which is the size of a USB key (www.nanoporetech.com). It contains biological pores through which DNA molecules pass. It is able to identify bases of DNA by measuring the changes they generate in electrical conductivity when the DNA strands flow through the pore. Sample preparation protocol includes the incorporation of a hairpin adapter that links the 2 strands of DNA molecule by one end. Both strands of a DNA molecule can be sequenced sequentially to generate a highly accurate consensus sequence. After numerous improvements of flow cells and sample preparation kits in 2014, latest released data showed that the MinION could deliver reads with a length up to 150 kilobases with an average of ~5 kilobases (Madoui et al. 2015). Some runs have produced up to 490 megabases of sequence in 48 h. The accuracy remains poor with an average identity (how closely the read matches a reference) of 75–85 % (Madoui et al. 2015). Nanopores are more than a single base in height so that the ionic signal measurements are not of individual nucleotides but of approximately 5 nucleotides at a time. Therefore, the base calling must individually recognize at least 45 = 1024 possible states of ionic current for each possible 5 mer, increasing dramatically the complexity of the signal. Two other nanopore-based sequencers are currently in development by Oxford Nanopore Technologies with increase throughput, the GridION, and the PromethION which are planned to generate 1 gigabase of sequence per minute.
Several other third-generation sequencers are currently in development, notably the GnuBIO system (Bio-Rad), NabSys sequencer, GeneReader (Qiagen), etc. Some of these systems should revolutionize sequencing as NGS did and consequently genomic scientific research as well as clinical genetic testing with very fast and cheap and reliable sequencing of long DNA pieces.
5.2.2 NGS Applications
5.2.2.1 Genomics
Recent progress in technology led to substantial cost reduction and increased throughput and accuracy of DNA sequencing. A flow of genetic data has continuously grown, and scientists across many fields have used NGS for a multitude of applications (Fig. 5.2). In genomics, sequencing and resequencing of full genomes require a lot of sequencing data but few preparation steps. DNA is extracted and sheared through mechanical or enzymatic action. The library preparation consists in end repair and adapter ligation. A human genome requires at least 100 gigabases of sequences, and smaller genomes such as Escherichia coli require as little as 125 megabases that represents a tiny fraction of the NGS throughput. Sequencing a whole genome is not a standard approach even today for research or clinical applications because of its associated cost despite a huge decrease over the last 7 years. For example, tumor samples are heterogeneous, and standard genome sequencing used for organism genomes does not produce enough data to have a clear picture of tumor-associated molecular events. The depth of coverage which represents the number of time any targeted base is sequenced by independent sequencing reads needs to be around 100×, whereas constitutive genomes are commonly sequenced with a depth of 30×. Therefore, comprehensive sequencing of a tumor genome would cost at least 3 times more.
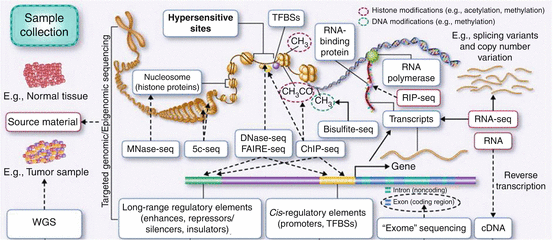
Fig. 5.2
NGS applications. WGS: whole genome sequencing; Mnase-seq: sequencing of nucleosome-associated DNA; 5c-seq (also 4C-seq, HiC-seq): chromosome conformation capture, identification of genome-wide regulatory interactions of a given locus with its unknown partners; DNAse-seq, FAIRE-seq: identification of open chromatin regions; ChIP-seq: identification of protein binding sites; Bisulfite-seq: identification of methylated regions, RIP-seq: identification of protein-RNA binding sites
An alternative is the sequencing of the exome, which represents 1 % of the genome and encompasses all coding regions, or the sequencing of several genes, one gene, or a part of gene. Such targeted sequencing is achieved through the enrichment of the region of interest during library preparation before sequencing. This strategy has a reduced cost compared to the genome and allows deeper investigation of the region of interest even if the targeting strategy could be expensive depending on the method. Enrichment of targeted regions is mainly performed following two different approaches, capture and PCR amplification (Mamanova et al. 2010). The capture method is mostly used for large targets such as the exome or more than 10 genes. After library preparation, library molecules are incubated with probes designed to hybridize with targeted genomic regions. After incubation, DNA-probe complexes are recovered usually by using streptavidin-coated beads that selectively bind the biotin linked to the probes. Thus, the sequenced libraries contain only the targets. A large cumulative size of targets can be sequenced through this method, but the capture of small targets often leads to a significant portion of off-target sequencing data. Main challenges of this method are the DNA’s high quality which could be challenging for some samples, long library preparation compared to other NGS applications, and specificity of the capture. Indeed, some thermodynamic constraints prevent from an efficient capture of some genomic regions (high or low GC content, repetitive regions) that lead to poor sequencing of these targets.
Isolation of regions of interest by PCR is usually the preferred method for small size cumulative target length. DNA is mixed with primers that are complimentary to regions of interest. Simultaneous amplification of all regions is performed with the multiplexing of all PCR reactions in one or more reaction tubes. Then, starting from amplified fragments, a library is prepared and sequenced. The main advantages of this technique are the ease of sample preparation and the low amount of DNA required. Furthermore, bad quality samples such as FFPE samples can be successfully processed by reducing the size of PCR products and increasing the number of primers. The main challenge of this approach is the uniformity of sequencing. Indeed, usually hundreds of PCR reactions simultaneously occur, depending on the size of the target, and since efficiency of each PCR reaction is usually nonequal, some targeted regions are poorly sequenced and some highly sequenced. This amplification heterogeneity amplifies along with the number of targets. The under- and over-sequencing has a huge impact on the final cost since the depth of sequencing wanted must be based on the poorest sequenced region. Similarly to capture method some challenging regions could neither be amplified nor sequenced. Other methods for targeted sequencing exist such as the Haloplex approach. It consists in the capture and amplification of targeted regions of DNA sheared through a constant pattern with specific cocktail of enzymes and using existing probes selected in a catalog. To help customers, exome and some predesigned gene panels have been optimized by manufacturers and can be purchased directly. Custom designs are set up through user-friendly web interface directly on manufacturer websites such as Agilent (SureSelect, Haloplex), Illumina (TruSeq, Nextera custom), and Thermo Fisher (AmpliSeq).
Related posts:
 Assessment of Biomarkers’ Predictive Value of Efficacy
Assessment of Biomarkers’ Predictive Value of Efficacy
 Introduction: Rationale for Precision Medicine Clinical Trials
Introduction: Rationale for Precision Medicine Clinical Trials
 The Key Role of Pathology in the Context of Precision Medicine Trials
The Key Role of Pathology in the Context of Precision Medicine Trials
 Microarrays-Based Molecular Profiling to Identify Genomic Alterations
Microarrays-Based Molecular Profiling to Identify Genomic Alterations
 Basis for Molecular Genetics in Cancer
Basis for Molecular Genetics in Cancer
 Bioinformatics for Precision Medicine in Oncology
Bioinformatics for Precision Medicine in Oncology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


