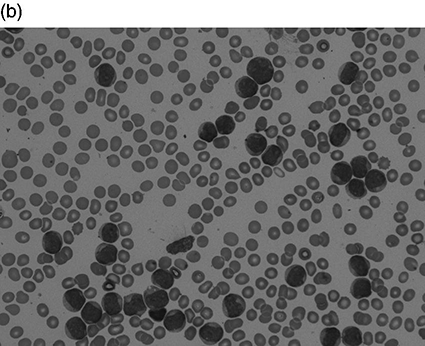
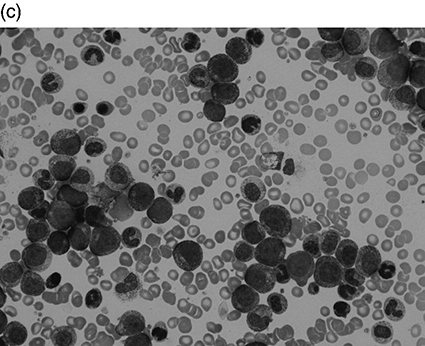
Chapter 4 Quentin A. Hill Department of Haematology, Leeds Teaching Hospitals NHS Trust, Leeds, UK Thrombocytosis, defined as a platelet count greater than 450 × 109/L, has been reported in 1–2% of patients attending hospital. It is a common finding in the intensive care unit (ICU), present in approximately 20% of patients at some stage during the admission [1–3]. The differential diagnosis of thrombocytosis can be seen in Table 4.1. As with unselected patients attending hospital [4], the vast majority of thrombocytosis cases are reactive in a critical care setting. Table 4.1 Causes of thrombocytosis. Circulating platelets are derived from bone marrow megakaryocytes. Thrombopoietin is the main regulator of platelet count, driving megakaryocyte maturation and proliferation. However, reactive thrombocytosis appears to be secondary to pro-inflammatory cytokines such as interleukins (especially IL-6) and TNF which are also known to affect thrombocytopoiesis. Patients may also have raised inflammatory markers and a leukocytosis. Severe illness can cause a left shift on blood film where myeloid stem cell precursors not normally seen in peripheral blood are visible. These include band forms, metamyelocytes, myelocytes and, less frequently the more immature forms, promyelocytes and myeloblasts. The blood film may reveal other features of inflammation such as rouleaux formation and reactive neutrophilia (see neutrophilia in the following text). If hyposplenism is the cause of thrombocytosis, additional supportive features may be found in the film (Howell–Jolly bodies, target cells, lymphocytosis and acanthocytes). In patients attending hospital, the most common causes are tissue damage, infection, iron deficiency, malignancy (most commonly gastrointestinal, lymphoma and lung) and chronic inflammatory disorders (most commonly inflammatory bowel disease, rheumatoid arthritis, chronic pancreatitis and systemic lupus erythematosus (SLE)) [4, 5]. Most critically ill trauma patients with thrombocytosis have additional predisposing conditions such as nosocomial infection, acute lung injury, bleeding and catecholamines [3], while in one study, independent risk factors for thrombocytosis included obesity, laparotomy, injury severity and mechanical ventilation [2]. The main concern with thrombocytosis is thrombosis. However, in the general hospital population, thrombosis only occurs in approximately 1% of patients with a reactive thrombocytosis, generally when additional risk factors (e.g. post-operative patient or underlying malignancy) are also present [4, 5]. Even at very high platelet counts, the risk appears low, and in a study of 231 patients with a reactive platelet count greater than 1000 × 109/L (sometimes labelled as extreme thrombocytosis), only 3 (1%) had vaso-occlusive symptoms/events [6]. Venous thromboembolic events (VTE) also appear infrequent in trauma patients with thrombocytosis admitted to the ICU (1/34 (2.9%) [3] and 28/614 (4.6%) [2]). In the series of Salim et al. [2], those with thrombocytosis had a significantly higher rate of pulmonary embolus compared to patients with normal platelet counts, but the overall rate of thrombosis was not significantly different. Thrombosis was no more likely in the 41 patients with a platelet count greater than 1000 × 109/L than those with platelets counts of 450–1000 × 109/L. Critically ill patients with thrombocytosis surprisingly have lower mortality compared to patients with normal counts [1–3], and individuals with a blunted platelet response (low rate of change following ICU admission), on the contrary, have higher mortality. The reason for this is not understood, but reactive thrombocytosis may be a marker of the host’s ability to successfully initiate a response to injury [2]. The onset of thrombocytosis is typically 7–14 days after ICU admission [1–3]. In one study, all patients’ platelet counts normalized with resolution of the acute illness at a mean of 35 days after admission [3]. Most primary thrombocytosis is caused by myeloproliferative neoplasms (MPNs) such as essential thrombocytosis (ET), polycythaemia vera (PV), primary myelofibrosis (PMF) and chronic myeloid leukaemia (CML). Thrombocytosis can also be due to myelodysplastic syndrome (MDS)/MPN overlap syndromes, e.g. chronic myelomonocytic leukaemia (CMML) and MDS with isolated del(5q-). Vasomotor symptoms such as headache, transient visual disturbance, chest pain, syncope and erythromelalgia (burning pain in hands or feet with erythema and warmth) occur in up to 40% of ET patients but are rare in reactive thrombocytosis [7]. Patients may also have splenomegaly, weight loss, night sweats, fatigue and loss of appetite. Other potential blood count abnormalities include a raised white count (e.g. CMML, CML), raised haematocrit (PV) or anaemia (PMF, MDS). Pseudohyperkalaemia can occur due to in vitro leakage of intracellular potassium during clotting. Thrombotic complications (mostly arterial) develop in 10–40% of those with ET and PV. Venous thrombosis usually occurs in the lower limbs but also in the abdominal veins (hepatic, portal or mesenteric) or the cerebral sinuses. Bleeding risk is also increased, especially when the platelet count is greater than 1000–2000 × 109/L. This has been attributed to platelet dysfunction and an acquired von Willebrand disease caused by large von Willebrand factor multimers being absorbed onto platelets. Definitive diagnosis usually requires a bone marrow aspiration and biopsy with cytogenetics. Peripheral blood testing for the JAK2V617F mutation is positive in approximately 50% of ET and PMF patients and over 90% of those with PV but is not found in reactive thrombocytosis. Treatment will depend on the exact diagnosis. ET patients typically receive aspirin, but platelets greater than 1000 × 109/L is a relative contraindication, requiring initial cytoreduction. Those at high risk of thrombosis (age > 60, prior ET-related thrombotic event) or haemorrhage (prior ET-related haemorrhage, platelet count >1500 × 109/L) will also receive cytoreductive therapy such as hydroxycarbamide, interferon or anagrelide. A blood film should be requested in all cases as this will exclude a pseudo-thrombocytosis (Table 4.1) and may detect features suggestive of a primary disorder such as macrocytosis, basophilic stippling, teardrop poikilocytes or a leukoerythroblastic picture. Other features suggesting a primary disorder include B symptoms, splenomegaly, vasomotor symptoms, and additional full blood count (FBC) abnormalities. Check inflammatory markers, but if there is an obvious secondary cause (e.g. acute blood loss, recent surgery/trauma, active infection or splenectomy scar), extensive investigation is not required. If no secondary cause can be found or the thrombocytosis preceded or persists despite resolution of the acute illness, haematology advice should be sought. Leukocytosis is defined as an elevation of the total white blood count (WBC) greater than two standard deviations above the mean (or a WBC > 11 × 109/L in adults). Leukocytosis is a common finding in critical care and is a marker of poor outcome in commonly used prognostic scoring systems. In most cases, the reason for leukocytosis is readily apparent, and treatment is of the underlying disorder. Instances when a more careful assessment is required are highlighted in the following text as well as the approach to, and differential diagnosis of, elevation in leukocyte subsets. Leukemoid reaction has been used to describe a marked reactive leukocytosis (sometimes defined as >50 × 109/L) with associated left shift. Common causes include severe bacterial infection, severe haemorrhage, tuberculosis, growth factors (e.g. granulocyte colony-stimulating factor (G-CSF)) and a paraneoplastic reaction to solid organ malignancy (the latter usually associated with large tumour burden and poor prognosis). Most importantly, leukemoid reaction must be distinguished from leukaemia. In leukemoid reaction, leukocytes are predominantly more mature cells, neutrophils typically have reactive changes (see neutrophilia in the following text), and there may be a reactive thrombocytosis. By contrast, in CML, the presenting WBC may be as high as 500 × 109/L with prominence of myelocytes and basophils, while splenomegaly can be massive (Figure 4.1). Hypogranular neutrophils, a predominance of blast cells and presence of Auer rods in blast cells favour leukaemia [10]. If leukaemia is suspected, send peripheral blood for immunophenotyping, but bone marrow examination with cytogenetics may be required. Figure 4.1 Leukocytosis. All are peripheral blood films at × 50 magnification. (a) Normal blood film. White blood count (WBC) 9 × 109/L. (b) Patient with acute myeloid leukaemia. Presenting WBC 197 × 109/L. (c) Patient with chronic myeloid leukaemia (CML). Presenting WBC 376 × 109/L (presented with blurred vision, retinal haemorrhage and massive splenomegaly).
High Blood Counts
Thrombocytosis
Primary
Inherited
Hereditary (e.g. mutations in gene for TPO or c-Mpl)
Acquired
Polycythaemia vera (PV)
Essential thrombocythaemia
Primary myelofibrosis (PMF)
Chronic myeloid leukaemia (CML)
Myelodysplastic syndrome (MDS)
Chronic myelomonocytic leukaemia (CMML)
Secondary
Infection (acute/chronic)
Inflammatory disorders
Tissue injury (including surgery)
Hyposplenism (surgical or functional)
Severe exercise
Severe haemolysis
Haemorrhage or iron deficiency
Malignancy
Drugs (steroids, adrenaline, vincristine, thrombomimetics)
Rebound post chemotherapy or alcohol withdrawal
Pseudo-thrombocytosis
Schistocytes (red cell fragments)
Microspherocytes (e.g. severe burns)
Cytoplasmic fragments (e.g. chronic lymphocytic leukaemia (CLL))
Bacteria
Haemoglobin H disease
Cryoglobulinaemia
Secondary (reactive) thrombocytosis
Primary thrombocytosis
Approach to thrombocytosis in critical care
Leukocytosis
Leukemoid reaction

Hyperleukocytosis and leukostasis
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


