Syndrome
Gene
Chromosome location
Mutation rate in sporadic RCC
Prevalence
% de novo
Renal tumor histology
Multiple/solitary
Penetrance of RCC
Age (years) at RCC diagnosis
Extrarenal manifestations
VHL disease
VHL
3p25
92% in sporadic ccRCC
1/36,000
20%
ccRCC
BMF
25–45%
45 (mean)
Pheochromocytoma
CNS hemangioblastoma
Retinal angioma
Endolymphatic sac tumor
Pancreatic tumor/cyst
Epididymal cystadenoma
Broad ligament cystadenoma
HPRC
MET
7q31
13% in papillary type 1 RCC
Very rare, around 30 families
47%
Papillary type 1 RCC
BMF
67%
46~63 (median)
None
HLRCC
FH
1q42
Not found in sporadic RCC
TBD
TBD
Papillary type 2 RCC (62.5%)
Mostly solitary
14–18%
39–46 (median)
Skin leiomyoma
Tubulopapillary (20%)
Tubular (5%)
Solid (1.5%)
Uterine leiomyoma
Mixed patterns (10%)
SDH-RCC
SDHB/C/D
SDHB:1p35–36 SDHC: 1q23 SDHD:11q23
Not found in sporadic RCC
TBD
TBD
Unique form of oncocytic RCC, ccRCC, chromophobe RCC
BMF
SDHB:14% SDHD:8%
SDHB:33 (mean) SDHC:47 (mean), SDHD:48 (median)
Pheochromocytoma
Paraganglioma
Papillary type 2 RCC, oncocytoma
BHDS
FLCN
17p11
Controversial (0~5% in chromophobe RCC)
TBD (report of 500 families worldwide)
TBD
Oncocytic hybrid tumor (50%), chromophobe RCC (35%), ccRCC (9%), oncocytoma (5%)
BMF
12–34%
48 (median)
Fibrofolliculoma, lung cyst, spontaneous pneumothorax
TSC
TSC1/TSC2
TSC1:9q34 TSC2:16p13
6% in chromophobe RCC, 2% in ccRCC
1/6000 to 1/10,000
66%–83%
Renal angiomyoadenomatous tumors (RAT)-like RCC
BMF
2–3%
30–42 (mean)
Angiofibroma fibrous cephalic plaque, ungual fibroma, shagreen patches, hypomelanotic macule, confetti-like macule, cortical dysplasia, subependymal nodule (SEN), subependymal giant cell astrocytoma (SEGA), cardiac rhabdomyoma, lymphangioleiomyomatosis (LAM), uterine PEComa
RCC with smooth muscle stroma, chromophobe-like RCC, unique granular eosinophilic-macrocystic RCC, TSC-associated papillary RCC, hybrid oncocytic/chromophobe tumor (HOCT), unclassified RCC, angiomyolipoma, epithelioid angiomyolipoma
Cowden syndrome
PTEN
10q23
7.5% in sporadic RCC, 9% in chromophobe RCC, 2–4% in ccRCC
At least 1/200,000
10.7%–47.6%
Papillary RCC, ccRCC, chromophobe RCC
Mostly solitary
34%
49 (median) 45 (mean)
Trichilemmoma (hair follicle hamartoma), papillomatous papule, acral/plantar keratoses, macrocephaly, dolichocephaly dysplastic gangliocytoma of the cerebellum, benign tumors(colorectal polyposis, thyroid goiter/nodule, lipoma, fibroma, and proliferative breast change), lifetime risk for cancer (breast cancer, thyroid cancer, endometrial cancer, colorectal cancer)
BAP1 cancer syndrome
BAP1
3p21
7.5 to 14% in ccRCC
TBD
TBD
ccRCC
BMF
TBD
45 (mean)
Malignant mesothelioma, uveal melanoma, cutaneous melanoma, melanocytic BAP-1 mutated atypical intradermal tumors (MBAITTs), lifetime risk for other cancers (breast cancer, lung cancer, neuroendocrine carcinoma, basal cell carcinoma, meningioma)
Through the study of hereditary RCC susceptibility syndromes, many important novel genes have been identified such as the VHL (von Hippel-Lindau) gene responsible for von Hippel-Lindau disease, and several previously known genes were rediscovered to have new essential functions including the MET proto-oncogene which is mutated in hereditary papillary renal cell carcinoma and FH (fumarate hydratase), the gene responsible for hereditary leiomyomatosis renal cell carcinoma. Cloning of these genes and elucidating their molecular genetics have contributed to a better understanding of the pathogenesis of these hereditary RCC susceptibility syndromes and to the development of specific genetic tests, appropriate surveillance, and targeted therapies. These studies have also provided insight into the molecular basis of non-hereditary, sporadic RCC. In this chapter, clinical manifestations and the genetics of hereditary RCC susceptibility syndromes and the molecular function of the responsible genes will be presented.
2.2 Von Hippel-Lindau (VHL) Disease
Von Hippel-Lindau (VHL) disease is an autosomal dominant hereditary neoplastic disorder and the first described hereditary kidney cancer syndrome. The clarification of the molecular pathogenesis of VHL disease has made an enormous contribution toward understanding the molecular mechanism of sporadic clear cell renal cell carcinoma (ccRCC) development and provided the basis for developing molecular targeted therapies for ccRCC.
2.2.1 Clinical Manifestations of Von Hippel-Lindau Disease
VHL disease is a rare disease which occurs about 1 in 36,000 [1, 2, 3], which is characterized by a predisposition to develop ccRCC, pheochromocytomas, central nervous system (CNS) hemangioblastomas, retinal angiomas, endolymphatic sac tumors, pancreatic tumors, cysts in the kidney and pancreas, epididymal cystadenomas, and broad ligament cystadenomas (Fig. 2.1a–i) [4–7]. VHL disease is classified generally into two subtypes, type 1 without pheochromocytoma and type 2 with pheochromocytoma. Type 2 is further subclassified into type 2A without RCC, type 2B with RCC, and type 2C with pheochromocytoma only without any other manifestations [8, 9]. Twenty-five to 45% of affected members of VHL families have bilateral, multifocal ccRCC [3]. Since the biological behavior of ccRCC in VHL disease is known to be mild and VHL patients have a lifetime risk for recurring ccRCC development, active surveillance is recommended until the size of the largest tumor reaches 3 cm in diameter. To conserve kidney function, nephron-sparing surgery including enucleation of the tumor is preferred as a surgical intervention [10, 11]. Pheochromocytomas develop in 10 to 20% of individuals with VHL disease, which can be multiple and bilateral. Extra-adrenal paragangliomas can arise in the carotid body and sympathetic paraganglia. Minor populations of pheochromocytoma in VHL disease can be malignant [3, 6]. CNS hemangioblastomas are the most common manifestations seen in 60–80% of affected patients. Although these are benign tumors, they are a major cause of morbidity in VHL disease because of their localization in the cerebellum, brainstem, and spinal cord [6, 12, 13].
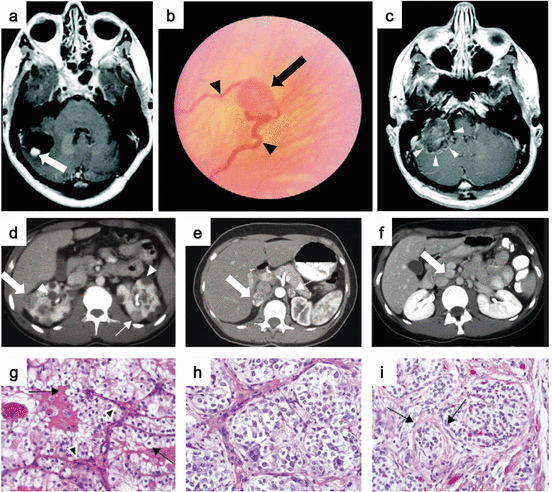
Fig. 2.1
Clinical manifestations of VHL disease. (a) MRI image of a cerebellar hemangioblastoma (arrow) with an associated cyst. (b) Ophthalmoscopic view of retinal hemangioblastoma (arrow) with an enlarged vessel (arrowheads). (c) MRI image of an endolymphatic sac tumor (arrowheads). (d) Postcontrast CT imaging shows bilateral multifocal RCC with solid (arrows) and cystic (arrowheads) disease. (e) Postcontrast CT image of bilateral pheochromocytomas (arrows). (f) Pancreatic neuroendocrine tumor (arrow). (g) Histology of clear cell RCC. (h) Histology of pheochromocytomas. (i) Histology of pancreatic neuroendocrine tumors with trabecular architecture (Images from Lonser et al. [6])
2.2.2 Genetics of Von Hippel-Lindau Disease
Loss of heterozygosity (LOH) on chromosome 3p was first found in sporadic RCC [14]. The study of age incidence for sporadic ccRCC and for RCC in VHL disease suggested that the chance of developing RCC in VHL disease was compatible with a “one-hit” model, while the chance of developing RCC in a sporadic setting was compatible with a “two-hit” model [15]. Based on these findings, a tumor suppressor for ccRCC was predicted to be located on chromosome 3p, and a novel VHL gene was isolated on chromosome 3p25–26 by positional cloning in VHL kindreds [16]. Individuals affected with VHL disease harbor a germline mutation in the VHL gene. LOH or somatic inactivation of the second allele was observed in VHL-associated RCC, indicating a classical tumor suppressor function for VHL [17, 18]. Germline VHL mutations in VHL disease encompass a broad spectrum of mutations, including frameshift mutations, nonsense mutations, large deletions, splicing defects, and missense mutations substituting an amino acid in the VHL protein. Over 945 VHL families worldwide have been analyzed for VHL germline mutations, and more than 700 different VHL mutations have been found throughout the entire VHL gene with the exception of the first 35 amino acids, which are not conserved across species [3]. VHL germline mutations were identified in nearly 100% of VHL families facilitated by the development of new methods to detect large deletions, confirming that VHL disease is caused solely by germline mutations in the VHL gene [19].
One of the major findings that has come from studying VHL disease to understand the molecular mechanism of RCC is that somatic mutations of the VHL gene accompanied by loss of the wild-type VHL allele are found in most sporadic ccRCC [20, 21]. Ninety-two percent of sporadic ccRCC are reported to have somatic mutations in or methylation of the VHL gene, indicating that loss of the VHL gene function is the fundamental initial step in most sporadic ccRCC development [22]. Insights gained from studies of families with VHL disease serve as a model for how discoveries obtained from study of a familial cancer may be applied to sporadic cancers.
2.2.3 Molecular Function of VHL Protein
The protein encoded by the VHL gene, pVHL, was a novel protein with no known functional domains, when it was isolated. Extensive research has clarified that pVHL functions as a substrate recognition component of an E3 ubiquitin ligase protein complex composed of elongin C, elongin B, Cul2, and Rbx1 [23–30]. Under normoxic conditions, transcription factors hypoxia-inducible factor 1α (HIF1α) and hypoxia-inducible factor 2α (HIF2α) are hydroxylated on their N-terminal transactivation domain (NTAD) by the EglN family of prolyl hydroxylases (PHDs), which require α-ketoglutarate, oxygen, ascorbic acid, and iron. Prolyl hydroxylated HIF1α and HIF2α are bound by the β-domain of pVHL, ubiquitinated and degraded by the proteasome [30–37]. Under conditions when oxygen or iron is insufficient, HIF1α/HIF2α is not hydroxylated, escapes from pVHL-mediated ubiquitination, and accumulates, driving transcription of hypoxia-responsive genes through their binding to hypoxia-responsive elements (HREs). Thus, in VHL-deficient cells, HIF1α/HIF2α is not ubiquitinated, and hypoxia-responsive genes, which are important for cell proliferation, including VEGF, PDGFB, TGFα, GLUT1, and CCND1, are upregulated even under normoxic conditions [38]. The fact that germline VHL mutations are frequently found in the α-domain of pVHL that interacts with elongin C and the pVHL β-domain, which interacts with prolyl hydroxylated HIFα, emphasizes the physiological importance of HIFα degradation for pVHL tumor suppressor function. HIF1α and HIF2α are similar in their structure, form heterodimers with HIFβ (ARNT) to bind to HREs, and share many hypoxia-responsive gene targets. However, their target genes are not identical and differ in a context-dependent manner. For example, glycolysis-related genes are mainly regulated by HIF1α, and CCND1 is regulated by HIF2α in RCC cells [38–43]. In terms of kidney cancer development, many in vitro and in vivo studies support the idea that HIF2α is a renal oncoprotein and HIF1α is a renal tumor suppressor [43–46]. Chromosome 14, where the HIF1α gene is located, is frequently deleted in ccRCC, and loss of 14q is associated with poor prognosis of ccRCC patients [47].
2.2.4 VHL Research: Bench to Bedside
As mentioned above, VHL disease research has made invaluable contributions to the clarification of the molecular mechanisms of ccRCC development and to the development of molecular target therapies for RCC [48]. Many drugs targeting the VHL-HIFα axis that have been approved by the FDA as therapeutic agents for advanced ccRCC patients have proven efficacy and superseded conventional immunotherapies. The details of targeted therapies for RCC will be discussed in other chapters.
2.2.5 Additional Gene Alterations in ccRCC
High throughput sequencing analysis of sporadic ccRCC has identified a number of gene alterations in addition to VHL mutations [49–51]. Genes mutated in sporadic ccRCC are involved in chromatin remodeling (PBRM1) [52], or histone modification, which regulates chromatin structure (SETD2, BAP1, JARID1C, and UTX, also known as KDM5C and KDM6A) [49, 53, 54]. Interestingly, PBRM1, SETD2, and BAP1 are located on chromosome 3p and could be deleted with VHL as a result of chromosome 3p loss. These gene alterations could contribute to ccRCC development and progression, which is initiated by loss of VHL. In fact, BAP1 mutation is associated with poor prognosis of ccRCC [51, 54]. Further analysis of the physiological consequence of alterations in these genes will provide a better understanding of the nature of ccRCC and might lead to the development of next-generation therapeutic agents for ccRCC.
2.3 Hereditary Papillary Renal Cell Carcinoma Type 1 (HPRC)
Hereditary papillary renal cell carcinoma type 1 (HPRC) is an autosomal dominant hereditary cancer syndrome (Fig. 2.2a), which was first described by Zbar et al. in 1994 [55, 56]. HPRC is a very rare type of hereditary RCC syndrome that predisposes affected individuals to develop bilateral multifocal papillary type 1 RCC (Fig. 2.2b) [57]. Causative germline mutations have been identified in the MET gene, which has an essential role in cancer cell proliferation, survival, invasion, and metastasis. Molecular genetic studies of HPRC have also contributed to our understanding of the molecular basis of RCC and provided the basis for development of targeted therapies for papillary RCC.
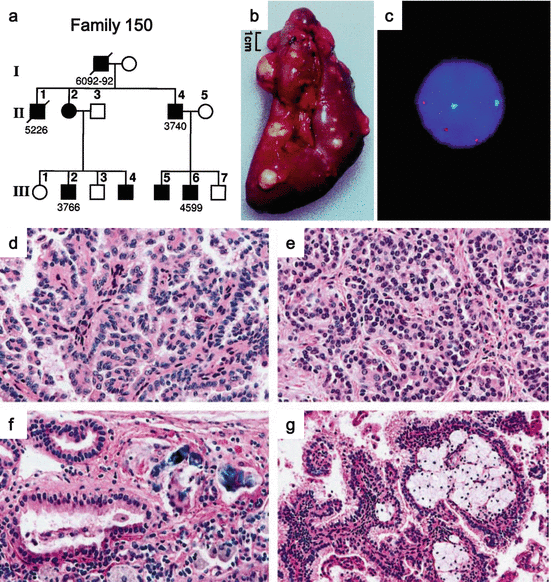
Fig. 2.2
Clinical manifestations of HPRC. (a) Pedigree of HPRC family. Solid symbols indicate individuals with RCC. (b) Gross image of a nephrectomized kidney from an HPRC patient. (c) Fluorescent in situ hybridization (FISH) on an RCC touch preparation shows trisomy of chromosome 7 (red chromosome 7, green chromosome 17). (d) Histology of RCC showing papillary architecture characterized by thin interstitium. (e) Tubulopapillary architecture composed of small RCC cells with basophilic nuclei and amphophilic cytoplasm. (f) Psammoma bodies are prominent histological features. (g) Most tumors in HPRC demonstrate foamy macrophages in fibrovascular cores. Focal clear cells can be seen occasionally (Images from Lubensky et al. [61])
2.3.1 Clinical Manifestations of HPRC
Distinct from other hereditary RCC syndromes, no manifestations other than RCC have been reported in HPRC. The patients have a lifelong risk for the development of multiple papillary type 1 RCC with age-dependent penetrance, which is estimated to be 67% by 60 years of age [58]. However, there are rare cases of HPRC kindreds presenting with earlier-onset RCC [59, 60]. RCC in HPRC tends to grow slowly, but is malignant and may metastasize when the tumor size becomes large. Since patients have a lifelong risk of developing multiple renal tumors, active surveillance is recommended until the largest tumor size reaches 3 cm when nephron-sparing surgery should be considered [11]. Histologically papillary type 1 RCC exhibits a characteristic papillary/tubulopapillary architecture lined by a single layer of small cells having small basophilic nuclei and amphophilic cytoplasm (Fig. 2.2d, e). Occasionally focal areas of cells with eosinophilic cytoplasm can be seen [61]. Fuhrman nuclear grade is predominantly 1–2. In some tumors, focal areas of Fuhrman nuclear grade 3 can be seen. Most tumors in HPRC exhibit foamy macrophages in fibrovascular cores. Psammoma bodies are frequently seen. There can be focal areas of clear cells (Fig. 2.2f, g). Multiple adenomas and microscopic papillary lesions can be seen in the renal parenchyma surrounding the tumors [61]. HPRC-associated RCC is hypovascular, and computed tomography (CT) imaging shows hypoenhancement with a contrast agent [57].
2.3.2 Genetics of HPRC
Trisomy of chromosome 7 was identified as a characteristic feature of papillary RCC, which suggested the localization of an oncogene on chromosome 7 [62, 63]. Through genetic linkage analysis in HPRC families, the responsible locus for HPRC was narrowed to chromosome 7q31.1–34, where the MET proto-oncogene was located. Schmidt et al. identified germline missense mutations in the tyrosine kinase domain of MET on chromosome 7q31 in affected individuals of HPRC kindreds [64]. Subsequently, somatic mutations of MET were identified in 13% of sporadic papillary type 1 RCC [64, 65]. These MET mutations were located in codons homologous to codons in KIT and RET, which were mutated in systemic mastocytosis and multiple endocrine neoplasia (MEN) type 2B, respectively. These findings support the idea that these missense mutations in MET are gain of function mutations, acquiring oncogenic activity.
2.3.3 Molecular Consequence of MET Mutation in HPRC
The MET proto-oncogene encodes c-Met, the hepatocyte growth factor/scatter factor (HGF/SF) receptor tyrosine kinase. HGF/SF, the ligand of c-Met, is produced by mesenchymal cells and stimulates a variety of neighboring cells including epithelial, endothelial, hematopoietic, and neuronal cells during normal embryonic development and throughout adulthood [66]. HGF/SF/c-Met signaling induces multiple biological activities, which include proliferation, survival, motility, epithelial-mesenchymal transition, and branching morphogenesis. Upon ligand binding, two tyrosine residues (Y1234 and Y1235) of c-Met in the activation loop of the tyrosine kinase domain are autophosphorylated and enhance c-Met kinase activity. Subsequent phosphorylation on two tyrosine residues (Y1349 and Y1356) near the carboxy terminus of c-Met form a multifunctional docking site which recruits a variety of signaling molecules, transmitting the signals further downstream for a variety of biological outputs [67, 68]. The pathological significance of MET missense mutations found in HPRC or PRC was investigated in NIH3T3 transfectants [69, 70]. Mutant c-Met showed increased autophosphorylation on tyrosine residues compared to wild-type c-Met. NIH3T3 cells expressing mutant c-Met are able to make foci on monolayer culture and form larger tumors in nude mice than cells expressing wild-type c-Met. In addition these cells displayed increased motility and increased activation of the Ras-Raf-MEK-ERK signaling pathway without HGF. Furthermore, the fact that a transgenic mouse model expressing mutant c-Met developed metastatic mammary carcinoma solidified the idea that mutant MET functioned as an oncogene [69, 70]. However, the data that support ligand-independent activation of mutant c-Met should be considered with caution. Most of the initial functional experiments with mutant c-Met were done in NIH3T3 cells, which express HGF/SF endogenously, but epithelial cells including renal tubular cells do not express HGF/SF. In fact, MDCK kidney epithelial cells reconstituted with c-Met mutants require the addition of exogenous HGF/SF for colony formation in soft agar. Deletion of the extracellular domain of mutant c-Met abrogates its transformation ability. In addition, expression of the soluble c-Met extracellular domain was able to block colony formation of NIH3T3 cells expressing mutant c-Met. Taken together, these data suggest that the availability of HGF/SF may contribute greatly to the oncogenesis of MET mutations in HPRC [71]. Mutant MET appears to have a lower threshold for kinase activation by HGF/SF, stabilizes the active conformation of the kinase, and exhibits a reduced susceptibility to inactivation by phosphatases in some cases [57]. It is noteworthy that most (95%) sporadic papillary type 1 RCC (PRC) exhibit chromosome 7 trisomy, while only 13% of PRC have somatic mutations in MET. Importantly, both MET and HGF/SF localize on chromosome 7. So trisomy 7 causes increased dosage of both HGF/SF and c-Met thereby driving HGF/c-Met signaling, which might be important for PRC development. Zhuang et al. precisely analyzed 16 RCCs in HPRC and found trisomy 7 in all tumors. Importantly, duplication of the specific chromosome 7 that harbors the mutant MET was seen in all 16 RCCs (Fig. 2.2c) [72]. This selective duplication of the mutant MET allele may function as a second hit event for RCC development in HPRC. These findings may suggest that the increased dosage of HGF/SF and c-Met and enhanced signaling through this axis is the essential factor for RCC development for both sporadic PRC and HPRC. Together with ligand dependency of mutant c-Met activation, these findings suggest an attractive hypothesis to explain why affected family members of HPRC develop cancer only in the kidney. The kidney produces large amounts of HGF/SF, as well as urokinase, which is necessary to activate the secreted immature form of HGF/SF [72].
2.3.4 HPRC Research: Bench to Bedside
These studies to understand the molecular pathogenesis of HPRC have provided significant insights into the development of targeted therapeutics [73]. Based on basic research, there are three possible strategies to target c-Met for HPRC and PRC: (1) direct inhibition of c-Met tyrosine kinase activity, (2) blockage of HGF/SF and c-Met interaction, and (3) inhibition of the molecular interaction between the cytoplasmic docking motif of c-Met and the effector downstream molecules. To date several humanized anti-HGF/SF monoclonal antibody drugs have been developed and are being tested in clinical trials for a variety of cancers [74]. An anti-c-Met humanized monoclonal antibody drug has also been developed and is being tested in a clinical trial for non-RCC cancers [75]. Small molecules targeting c-Met kinase activity are also being tested for efficacy in treating PRC and HPRC. The presence of germline mutations in MET is a factor well correlated with a positive response [75–77]. Since c-Met is activated in VHL-deficient ccRCC cells, c-Met could also be a target molecule for advanced ccRCC therapy [78].
2.4 Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
HLRCC is an autosomal dominant hereditary kidney cancer syndrome that was first reported in 2001 [79] as an inherited susceptibility to uterine leiomyomas and papillary RCC. HLRCC is caused by germline mutations of the fumarate hydratase (FH) gene encoding the TCA cycle enzyme [80]. RCC in HLRCC is very aggressive and has to be managed totally differently from RCCs that develop in other types of hereditary kidney cancer syndromes.
2.4.1 Clinical Manifestations of HLRCC
HLRCC is characterized by three manifestations: cutaneous leiomyomas, uterine leiomyomas (fibroids), and renal tumors and benign renal cysts. Originally HLRCC was reported in 1973 as Reed’s disease, in which patients presented with cutaneous leiomyomas and uterine leiomyomas. Subsequently cosegregation of kidney cancer with skin and uterine leiomyomas was identified, and Reed’s disease was renamed HLRCC (Fig. 2.3a). Skin leiomyomas are the most common manifestations seen in 76–100% of affected individuals with HLRCC, which present as multiple, firm, skin-colored to light-brown-colored papules and nodules (Fig. 2.3b) [81–83]. The number of lesions range from 10 to 100, and the size ranges from 0.4 to 2.5 cm in diameter. They develop on the trunk and extremities increasing over time with a disseminated pattern or a combination of disseminated and segmental distribution. Most of the lesions are symptomatic with pain and paresthesias. Mean age of onset of cutaneous lesions is 25 years (range 10–47 years old) [81]. The other common manifestations are uterine leiomyomas (fibroids), developing in most women affected with HLRCC (Fig. 2.3c) [81–85]. Multiple leiomyomas with diameters ranging from 1.5 cm to 10 cm develop very early (median 28–31 years) in HLRCC patients [84, 86]. In one study, 91% of affected women with skin and uterine leiomyomas had a myomectomy or hysterectomy, and 57% of affected women with skin and uterine leiomyomas had a hysterectomy before 30 years of age [81]. Uterine leiomyomas in HLRCC show characteristic histology including increased cellularity, large single nuclei, or multiple nuclei with large orangiophilic nucleoli surrounded by a halo [86].
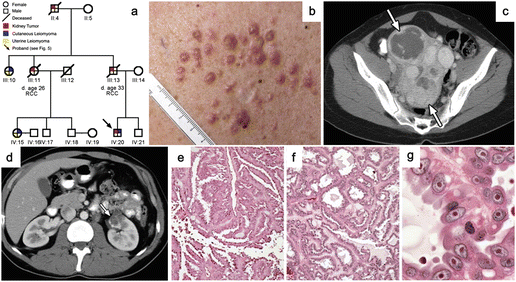
Fig. 2.3
Clinical manifestations of HLRCC. (a) Pedigree of an HLRCC family. Red quadrants indicate individuals with RCC. (b) Multiple skin leiomyomas on the trunk. (c) CT image of multiple large uterine leiomyomas (arrows). (d) CT image of a solitary RCC. (e) Histology of RCC showing papillary type 2 RCC architecture with thick and elongated collagen-abundant stalks. (f) Other architectural patterns including tubulopapillary are seen. (g) The characteristic large nucleus with a prominent large inclusion-like eosinophilic or orangiophilic nucleolus surrounded by a clear halo (Images from Grubb et al. [89])
HLRCC patients have an increased risk for developing RCC compared to unaffected family members. However, the penetrance for RCC in HLRCC is much lower than for leiomyomas, ranging from 14 to 18% in North American and French studies, and even lower in a Dutch study [87, 88]. In contrast to other hereditary kidney cancer syndromes, most RCCs in HLRCC are solitary and unilateral (Fig. 2.3d). However, there are reports of two cases of bilateral or bilateral, multifocal RCC among 38 HLRCC patients [89, 90]. RCC can develop in HLRCC at a young age (10–44 years) [79, 81, 89] with a reported median age at diagnosis of 39–46 years [81, 89]. The histology of HLRCC-related RCC is classified typically as papillary type 2 RCC, in which papillae are thick and elongated with fibrovascular cores (Fig. 2.3e). Many RCCs in HLRCC show this papillary pattern (62.5%), which is composed of characteristic cells with abundant amphophilic cytoplasm and large nuclei (Fig. 2.3g). However, it should be noted that they may also display other architectural patterns including tubulopapillary (20%) (Fig. 2.3f), tubular (5%), solid (1.5%), and mixed patterns (10%) [90]. Many cases of RCC in HLRCC have no cystic component (47.5%), but some cases have cystic areas (40%) or are predominantly cystic (12.5%). The hallmark of the HLRCC tumors is the presence of a characteristic large nucleus with a prominent large inclusion-like eosinophilic or orangiophilic nucleolus surrounded by a clear halo (Fig. 2.3g). Although the histology can be variable in HLRCC tumors, the characteristic feature of a macronucleus and prominent nucleolus with clear halo is commonly seen in all RCC in HLRCC [90]. Based on the nucleolus size, Fuhrman nuclear grade is classified as high grade in all cases. To date, there is no specific immunohistochemical marker for HLRCC-associated RCC. HLRCC-related RCC reveals an extremely malignant character, which differentiates HLRCC from other hereditary kidney cancer syndromes. More than 70% of RCC patients with HLRCC present with advanced stage III or stage IV disease [79, 81, 89]. Importantly, HLRCC-related RCC tends to metastasize to lymph nodes very early, when the primary tumors are T1. Even if patients initially present with localized disease, 50% will eventually develop lethally metastatic disease [89]. Based on this malignant nature, HLRCC-related RCC must be treated differently from other inherited forms of RCC. HLRCC tumors should be surgically treated immediately upon detection regardless of size rather than management by active surveillance until the largest tumor size reaches 3 cm, which is recommended for most other inherited RCC syndromes [81, 91, 92].
2.4.2 Genetics of HLRCC
Genetic linkage analysis in HLRCC families localized the disease locus on chromosome1q42, and germline mutations were identified in a gene encoding fumarate hydratase (FH), an enzyme of the tricarboxylic acid (TCA) cycle, which catalyzes the conversion of fumarate to malate [80, 81]. These mutations are predicted to cause absence or truncation of the FH protein or substitutions or deletions of conserved amino acids. Missense mutations are most common. Moreover FH enzyme activity was shown to be absent or reduced in tumors, lymphoblastoid cells, and fibroblasts from HLRCC patients [80, 82, 94, 95]. LOH studies show loss of the wild-type FH allele in skin and uterine leiomyomas and RCC, indicating FH is a classical tumor suppressor gene in HLRCC. More than 150 unique FH mutations, which may be pathogenic, have been reported in the Leiden Open Variant Database [96]. The mutation detection rate in affected individuals with HLRCC is reaching 90% [81, 82, 95]. There is a missense mutational hot spot at Arg190, which is mutated to histidine, leucine, or cysteine [81, 82, 95]. Kiuru et al. searched for FH mutations in sporadic skin and uterine leiomyomas and sporadic RCC and found few mutations [97].
2.4.3 Molecular Consequence of Mutation in FH
FH functions as a tetramer. Reduced FH enzymatic activity in lymphoblastoid cells and fibroblasts from HLRCC patients indicates that mutant FH may function as a dominant negative form to disturb normal function of the FH tetramer [98, 99]. Loss of FH activity impairs oxidative phosphorylation enabling a cell metabolism shift to aerobic glycolysis [100, 101]. Due to a blockage in the TCA cycle, accumulated fumarate and succinate are transported out of the mitochondria into the cytoplasm and compete with α-ketoglutarate, which is a cosubstrate of the EglN family of prolyl hydroxylases (PHDs) that target HIFα , resulting in inhibition of PHD activity and accumulation of HIFα and thereby evading pVHL-mediated ubiquitination and proteasomal degradation [102, 103]. This pseudo-hypoxic condition results in elevation of HIF-target genes such as VEGF and GLUT1, which leads to upregulated angiogenesis and glucose uptake [104]. More evidence supporting the oncometabolite function of fumarate and succinate is accumulating. Elevated fumarate and succinate in FH-mutated RCC can inhibit multiple α-ketoglutarate-dependent dioxygenases, which include histone demethylases (KDMs, JMJDs), prolyl hydroxylases, and the ten-eleven translocation (TET) family of DNA hydroxylases. As a consequence of dioxygenase impairment, genome-wide epigenetic alterations could occur which may contribute to kidney cancer development in HLRCC patients [105]. RCCs in HLRCC have increased levels of ROS, leading to HIFα stabilization [101]. This suggests another mechanism of tumor suppression by FH as well as the possible involvement of the antioxidant response in RCC development in HLRCC. S-(2-succinyl) cysteine (2SC) has been identified as an endogenous chemical modification of proteins. Fumarate is an electrophile and reacts with cysteine sulfhydryl groups to form 2SC under physiological conditions [106]. This reaction is termed succination, which could be detected endogenously, and modifies the activity of many proteins [107]. One of the significant proteins that is modified by succination is KEAP1. KEAP1 is the substrate recognition subunit of an E3 ubiquitin ligase complex, which is composed of KEAP1, Cul3, and Rbx1. This complex ubiquitinates a transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) for proteasome mediated degradation [108, 109]. Nrf2 transcriptionally upregulates target genes containing antioxidant-response elements (ARE), in response to oxidative and electrophilic stress [110]. In FH-mutated cells, elevated fumarate causes succination of critical cysteine residues in KEAP1, resulting in a conformational change of the KEAP1 containing E3 complex and an abrogation of Nrf2 recognition by KEAP1. As a result, Nrf2 is stabilized, accumulates, and upregulates ARE-containing genes leading to overactivation of the Nrf2-dependent antioxidant pathway [111–113]. In fact, somatic mutations in NRF2 and CUL3 are found in sporadic type 2 papillary RCC, which result in Nrf2 activation. Consistent with this model, loss of function mutations in KEAP1 are frequently seen in sporadic cancers [114–119]. Although Nrf2 is activated to transcriptionally upregulate antioxidant genes, ROS levels are still high in FH-deficient RCC cells. Sullivan et al. found that accumulated fumarate directly binds the antioxidant glutathione (GSF), which works as an alternative substrate to glutathione reductase, resulting in decreased NADPH and increased ROS [120].
2.4.4 HLRCC Research: Bench to Bedside
HLRCC-associated RCC has very malignant features and metastasizes even when the primary tumor is small in size. Advanced RCC in HLRCC is refractory to conventional immunotherapy and lethal. However, results from many research studies have been reported in the decade since FH germline mutations were identified in HLRCC, which may provide a foundation for the development of rational targeted therapies. Based on basic research, a phase II clinical trial to evaluate combination therapy of bevacizumab (anti-VEGF monoclonal antibody) and erlotinib (EGFR inhibitor) is currently under way at the National Cancer Institute, NIH (https://clinicaltrials.gov/ct2/show/NCT01130519?term=HLRCC&rank=1). The recent findings that define the KEAP1-Nrf2 axis in HLRCC may provide another basis for developing new targeted therapies for advanced HLRCC-associated RCC.
2.5 Hereditary Head and Neck Paragangliomas (HPGL) and Pheochromocytomas (PCC): SDH-RCC
Hereditary head and neck paragangliomas (HPGL), extra-adrenal pheochromocytomas (paragangliomas), and hereditary pheochromocytomas (PCC) are caused by germline mutations in genes encoding the three subunits (SDHB, SDHC, and SDHD) of the mitochondrial TCA cycle enzyme succinate dehydrogenase (SDH) [121, 122]. Bilateral multifocal RCC was reported as a novel manifestation of SDHB-mutated HPGL in 2004 [123]. A unique form of oncocytic RCC is seen most frequently in SDH-RCC. However, a variety of histologies including clear cell RCC, chromophobe RCC, papillary type 2 RCC, and oncocytoma have been reported [124–127]. SDH-RCC also can be very aggressive, similar to HLRCC.
2.5.1 Clinical Manifestations of SDH-RCC
Fifty-four percent of affected individuals with SDHB germline mutations developed HPGL or PCC and 79% of SDHC mutation carriers presented with HPGL or PCC. PCC and HPGL can be bilateral and/or multifocal [125, 128]. The mean age of diagnosis is younger for PCC (42.3 and 40.1 years of age) than for HPGL (27.4 and 20.7 years of age) in SDHB and SDHD mutation carriers, respectively. Approximately 13.6% of SDHB mutation carriers and 3.2% of SDHD mutation carriers developed malignant PCC or HPGL [126]. The frequency of RCC development is not very high. The lifetime risk of developing a renal tumor at the age of 70 was 14% in SDHB and 8% in SDHD mutation carriers, respectively [126]. RCC associated with SDH-RCC can have bilateral, multifocal, and early-onset characteristics.
2.5.2 Genetics of SDH-RCC
Germline mutations in SDHD were first identified in HPGL families in 2000 [121]. An SDHD germline mutation was found in a kindred with familial PCC as well [129]. Subsequently, germline mutations in SDHB and SDHD were also identified as causes of susceptibility to familial PCC and HPGL [130, 131]. In 2004, two young affected family members with HPGL and germline SDHB mutations were diagnosed with clear cell RCC [123]. Subsequently RCCs with a variety of histologies have been identified in family members inheriting germline mutations in SDHB, SDHC, and SDHD (Fig. 2.4a–d) [124–128]. All types of loss of function germline mutations including missense, frameshift, and nonsense are seen in SDH-RCC kindreds.
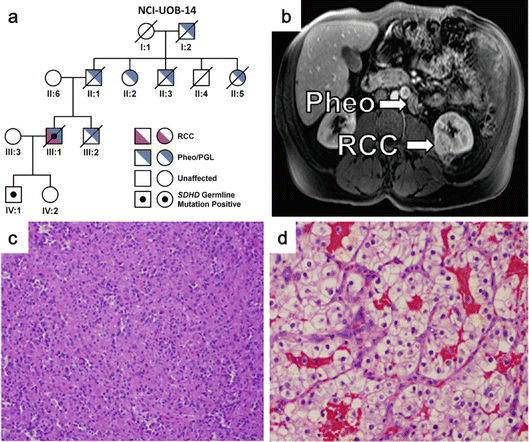
Fig. 2.4
Clinical manifestations of SDH-RCC. (a) Pedigree of an SDHD mutation-associated SDH-RCC family. Patient III:1 had advanced ccRCC. (b) MRI image of an SDHB mutation-associated SDH-RCC patient showing a pheochromocytoma and an RCC. (c) A unique form of oncocytic RCC is seen most frequently in SDH-RCC. (d) Histology of ccRCC seen in an SDHC mutation-associated SDH-RCC patient (Images from Ricketts et al. [127])
2.5.3 Molecular Consequence of Mutations in SDHB, SDHC, and SDHD
SDH enzymatic activity is impaired in SDH-mutated cells, resulting in the accumulation of succinate. Similar to increased fumarate in FH-deficient cells in HLRCC, accumulating succinate is exported into the cytoplasm and can compete with α-ketoglutarate, resulting in inhibition of enzymes which utilize α-ketoglutarate as a cosubstrate, including prolyl hydroxylases (PHDs) [102, 103, 105]. Upon PHD inactivation, HIFα evades ubiquitination by the pVHL E3 complex, accumulates, and transcriptionally activates expression of HIFα target genes that support cell proliferation through neovascularization, glucose uptake, or cell proliferation. Analogous to FH-mutated HLRCC, RCCs in SDH-RCC tend to have malignant features [127]. Based on the molecular consequence of SDH inactivation that drives upregulation of HIFα target genes, targeted therapies such as anti-VEGF antibodies or VEGFR inhibitors are expected to be effective for advanced RCC associated with SDH-RCC. In fact, there is a case report of advanced RCC in SDH-RCC, which shows nearly complete remission in response to a standard regimen of sunitinib [132].
2.6 Birt-Hogg-Dubé Syndrome (BHDS)
Birt-Hogg-Dubé syndrome (BHDS) is an autosomal dominant hereditary kidney cancer syndrome, which predisposes affected individuals to develop benign tumors of the hair follicle (fibrofolliculomas), pulmonary cysts, spontaneous pneumothorax, and kidney tumors (RCC and/or oncocytoma) (Fig. 2.5a–d). Causative germline mutations were identified in a novel gene FLCN in affected BHDS family members. In contrast to other hereditary kidney cancer syndromes, a variety of histologies including chromophobe RCC, oncocytoma, ccRCC, papillary RCC, and hybrid tumors consisting of features of both chromophobe RCC and oncocytoma can be seen in BHDS.
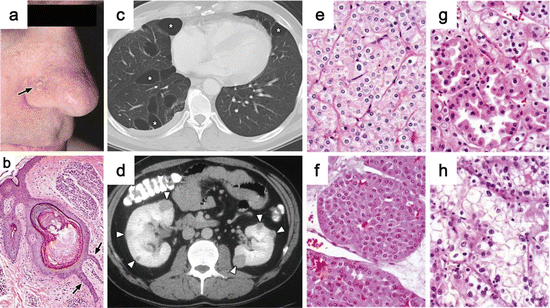
Fig. 2.5
Clinical manifestations of BHDS. (a) Fibrofolliculomas on the face (arrow). (b) Histology of fibrofolliculoma showing epithelial strands with thick connective tissue stroma (arrows). (c) CT image indicating multiple lung cysts. (d) CT image of bilateral multifocal renal tumors (arrowheads). (e–h) BHDS-associated renal tumors show multiple histological types: chromophobe RCC (e), oncocytoma (f), hybrid oncocytic tumor (g), and ccRCC (h) (Images from Pavlovich et al. [146])
2.6.1 Clinical Manifestations of BHDS
BHDS was first described in 1977 by three dermatologists, Birt, Hogg, and Dubé, as a hereditary cutaneous disorder in which patients presented with fibrofolliculomas [133]. A case report of a BHD patient having bilateral multifocal chromophobe RCC in 1993 raised the question of whether kidney tumors might be part of the manifestations of BHDS. One hundred fifty-two patients from 49 familial renal tumor families were analyzed for cutaneous lesions at the National Institutes of Health in the U.S. The cosegregation of fibrofolliculomas and kidney tumors was seen in three families in an autosomal dominant manner, which established BHDS as a hereditary kidney cancer syndrome [134]. BHDS is a rare syndrome with roughly 500 families reported worldwide to date. However, this number could be underestimated because BHDS is a newly categorized syndrome and not widely known yet.
Fibrofolliculomas are the most common clinical manifestations of BHDS, which are seen in 82–92% of affected individuals with BHDS who are older than 25 years old. It is a benign tumor, so-called hamartoma, seen as flesh-colored papules with a smooth surface, 2–4 mm in diameter, and frequently seen on the face, neck, and upper trunk singly or coalescing into a plaque (Fig. 2.5a) [135–138]. Histologically, fibrofolliculomas show anastomosing epithelial strands emanating from an aberrant hair follicle, surrounded by a thick fibrous tissue and mucin-rich stroma (Fig. 2.5b) [133, 134]. Other than a cosmetic issue, fibrofolliculomas exhibit no symptoms.
Lung cysts are the second most common manifestations of BHDS. Multiple bilateral thin-walled lung cysts can be observed on thin-section chest CT scans in 70–84% of affected individuals with BHDS (Fig. 2.5c) [135, 136, 138] Tobino et al. precisely described the characteristic features of pulmonary cysts in BHDS seen on CT scans. The cysts vary in their numbers (29–407/person), sizes (a few mm–2 cm or larger), and shape (76.6% of cysts are irregular-shaped). Cysts are predominantly distributed to the lower, medial, and subpleural regions of the lung, abutting or involving the proximal portion of lower pulmonary arteries or veins [139]. Respiratory function tests generally exhibit normal lung function [139, 140]. Affected family members with BHDS have a 50-fold higher risk for having spontaneous pneumothorax than unaffected siblings [141]. An analysis of 198 patients from 89 BHDS families evaluated for risk of pneumothorax revealed that 24% of BHDS patients had a history of pneumothorax. The presence of lung cysts, total lung cyst volume, and largest cyst size were significantly associated with pneumothorax. The median age of onset for pneumothorax in BHDS is 38 years old [142].
Zbar et al. performed a risk assessment study of a large cohort of BHDS families and concluded that affected family members of BHDS have a sevenfold greater risk of developing renal tumors than unaffected siblings [141]. The penetrance of renal tumors in BHDS ranges from 12 to 34% [135, 136, 143]. Kidney neoplasia present in BHDS patients can be bilateral, multifocal, or solitary (Fig. 2.5d). Kidney tumors in BHDS exhibit a wide spectrum of histological subtypes both in the single kidney of a BHDS patient and in multiple affected individuals from the same BHDS kindred, differentiating this syndrome from other hereditary kidney cancer syndromes (Fig. 2.5e–h). The unique kidney tumors, hybrid oncocytic/chromophobe tumors, which contain features of chromophobe RCC and renal oncocytoma [144] are the most common kidney tumors in BHDS (Fig. 2.5g). Pavlovich et al. have reported the frequency of histologies seen in BHDS-related kidney tumors as follows: hybrid oncocytic/chromophobe tumors (50%), chromophobe RCC (35%), clear cell RCC (9%), and renal oncocytoma (5%) [145, 146]. Multiple microscopic foci of eosinophilic dysplastic cells, so-called oncocytosis, can be seen frequently in the normal parenchyma of kidneys from BHDS patients [145]. Kidney tumors in BHDS tend to grow slowly and less aggressively, although they have the potential to metastasize. Affected family members without renal masses are recommended to be screened for kidney tumors by MRI every 36 months starting at the age of 21. If a renal mass less than 3 cm is detected, annual or semiannual imaging, depending on the size, location, and growth rate, should be considered. When the diameter of the largest tumor reaches 3 cm, surgical intervention is recommended. Since BHDS patients have a lifelong risk for developing multiple bilateral renal tumors, nephron-sparing surgery should always be considered to conserve renal function as much as possible, to prepare for multiple surgeries. During the nephron-sparing surgery of the largest tumor, all of the detectable small tumors should be removed with the aid of intraoperative ultrasound [146, 147]. So far there is no report of metastatic RCC developing in BHDS patients with primary tumors less than 3 cm in diameter [11]. However, BHDS-associated large RCC can metastasize and cause mortality [146, 148]. Appropriate regular follow-up has to be performed for BHDS-related kidney tumors.
Although it is not clear whether or not they are real BHDS manifestations, there are many reports of neoplasms in BHDS patients. Parotid oncocytomas have been identified in many BHDS patients [135, 136, 149–151]. It is controversial whether BHDS patients are at risk of developing colon polyps and/or colorectal carcinoma. There are several case reports describing colorectal manifestations in BHDS patients [152–154]. However, Zbar et al. have conducted a risk assessment study of BHD families who were evaluated by colonoscopy and showed no increased risk for colon polyps and/or carcinomas in affected members of BHDS families compared to unaffected members. On the other hand, Nahorski et al. and Khoo et al. have described increased risk of colorectal neoplasia in a large European BHD cohort and in a large French BHD family, respectively [155, 156]. Further analysis will be required to conclude if colon neoplasia should be included as a manifestation of BHDS.
2.6.2 Genetics of BHDS
The causative gene for BHDS was localized on the short arm of chromosome 17 by genetic linkage analysis in BHDS kindreds [157–159]. Subsequently in 2002, germline mutations were identified in a novel gene in affected family members with BHDS, which was named FLCN [160]. Since the cloning of FLCN, many germline mutations have been reported [136, 137, 144, 162, 163]. To date more than 100 unique germline mutations have been reported in the Leiden Open Variation Database (LOVD) for FLCN [163] (https://grenada.lumc.nl/LOVD2/shared1/home.php?select_db=FLCN). Lim et al. have reported analysis of 70 unique germline mutations based on this database in 2010. Germline mutations are found in all coding exons (4–14). Deletion mutations are most frequently seen (31/70, 40%), followed by single base substitutions (25/70, 35.7%), duplications (10/70, 14.3%), and deletion/insertions (4/70, 5.7%). Most of these germline mutations are predicted to cause loss of function of the encoded protein FLCN. Frameshifts, causing a premature termination codon, are the most frequent mutational consequences (37/70, 52.9%), followed by splice site mutations (14/70, 20%), nonsense mutations (10/70, 14.3%), missense mutations (6/70, 8.6%), and deletion mutations (3/70, 4.3%) [163]. Partial gene deletions of FLCN have been seen in the germline of affected BHDS family members, which are also predicted to cause loss of function [143, 161, 164]. Benhammou et al. have reported germline intragenic deletion of the noncoding exon 1 causing loss of promoter activity of the FLCN gene [164]. The mutation detection rate of FLCN in affected BHDS patients is reaching 90% with advanced technologies for identifying gene deletions and accurate sequencing [135, 136, 143]. There is no report of a clear genotype-phenotype correlation in BHDS [135, 136, 142].
The majority of tumor suppressor genes that are causative for hereditary cancer syndromes follow the Knudson two-hit theory. Tumors have germline loss of function mutations in one allele and additional inactivation of the other allele by LOH, somatic mutation, or methylation [165]. A second hit somatic inactivation of FLCN is seen in BHD associated renal tumors [156, 166]. Vocke et al. analyzed 77 renal tumors from 12 individuals with BHDS who were confirmed to carry germline mutations in FLCN. The majority of renal tumors (41/77, 53%) showed somatic mutations in FLCN, most of which resulted in frameshifts and loss of function. LOH at the FLCN locus was also seen at a relatively lower frequency (14/77, 17%). Interestingly, each tumor within a group of multifocal tumors from a single kidney of a BHDS patient showed a distinct second hit inactivation [166]. These observations support the idea that FLCN is a classical tumor suppressor gene, which follows the Knudson two-hit theory.
FLCN somatic mutations are seen infrequently in sporadic RCC. Multiple losses of whole chromosomes are a characteristic of sporadic chromophobe RCC. Chromosome 17, where FLCN is located, is frequently lost in chromophobe RCC [167]. This motivated Gad et al. to look at the somatic mutations of FLCN in sporadic renal tumors including 46 samples of chromophobe RCC, 19 ccRCC, 18 renal oncocytoma, and 9 papillary RCC. After five samples of chromophobe RCCs having mutations in normal tissues were excluded, somatic FLCN mutations were seen in 4.9% of sporadic chromophobe RCCs and in 5.6% of sporadic renal oncocytoma. No FLCN mutations were seen in ccRCC or papillary RCC. Methylation status of the FLCN promoter was analyzed in 61 of 92 samples, and no FLCN promoter methylation was found [168]. Khoo et al. analyzed 39 renal tumors, 7 samples of renal oncocytomas, 9 chromophobe RCC, 11 papillary RCC, and 12 ccRCC. Only one papillary RCC exhibited a somatic frameshift mutation in FLCN. However, LOH on chromosome 17p was observed in 36% of sporadic renal tumors (33% of all chromophobe RCC), and FLCN promoter methylation was detected in 28% of sporadic renal tumors (36% of all chromophobe RCC). Interestingly, 11% of chromophobe RCC, 27% of papillary RCC, and 8% of ccRCC showed both LOH and promoter methylation [169]. On the other hand, da Silva et al. found no evidence of FLCN CpG island methylation in 20 RCC tumors and 6 RCC cell lines. Nagy et al. did not find FLCN somatic mutations in any of 8 sporadic chromophobe RCC or 8 sporadic renal oncocytoma. They saw LOH on chromosome 17p in 100% of chromophobe RCC and 0% of oncocytoma [170]. The latest publication from the Cancer Genome Atlas project describing whole-exome sequencing of 66 sporadic chromophobe RCCs reports no mutations in FLCN [171].
2.6.3 Molecular Function of FLCN
FLCN gene encodes a novel 579 amino acid protein FLCN, which does not share any homology or known functional domains with other proteins at the level of amino acid sequence or secondary structure prediction [160]. However, FLCN is well conserved across species, suggesting its fundamental role for organisms. Baba et al. have identified a novel FLCN-binding protein, FNIP1, which is also conserved across species and has no known functional domains to suggest its function. FNIP1 binds to the C-terminus of FLCN, which is sometimes the target of protein truncating germline FLCN mutations in BHDS families, and interacts with 5′-AMP-activated protein kinase (AMPK), which has an important role as an energy sensor and metabolic switch to maintain energy homeostasis in cells and organisms [172, 173]. AMPK negatively regulates mechanistic target of rapamycin (mTOR) [174], the master regulator of protein translation and cell growth [175]. The significant role of the AMPK-mTORC1 signaling axis is well documented in hereditary cancer syndromes [176] including Cowden syndrome which is caused by PTEN inactivation [177], Peutz-Jeghers syndrome caused by LKB1 inactivation [178, 179], and tuberous sclerosis complex caused by TSC1 or TSC2 inactivation [180]. Indeed, there are several lines of evidence supporting FLCN/FNIP1 involvement in the AMPK-mTORC1 signaling pathway. FLCN is phosphorylated on multiple serines and threonines, which are differently inhibited by mTORC1 inhibition or AMPK inhibition. FNIP1 expression facilitates FLCN phosphorylation in an mTORC1 dependent manner [181, 182]. Regulation of mTORC1 activity by FLCN/FNIP1 seems to be context dependent. For example, a FLCN-null RCC cell line showed higher mTORC1 activity than the FLCN-restored RCC cell line under serum-starved conditions. On the other hand, serum stimulation activated mTORC1 inefficiently in the FLCN-null RCC cell line under amino acid-starved conditions, while the FLCN-restored RCC cell line demonstrated efficient activation of mTORC1 [181]. Recently, Tsun et al. have shown that FLCN functions as a RagC/D GTPase-activating protein (GAP) to facilitate mTOR recruitment to the lysosome for amino acid-dependent mTORC1 activation. Petit et al. have also shown that FLCN is required for mTOR to be recruited to lysosome by Rags upon amino acid stimulation. In this case they showed that FLCN functions as a RagA guanine nucleotide exchange factor (GEF) [183]. A crystal structure of the C-terminal half of FLCN was solved in 2012 and found to be structurally similar to a Rab-GEF family of proteins [184]. Animal models also support a complex FLCN role in mTOR regulation. Kidney-targeted Flcn deletion causes acute cell proliferation in kidney epithelial cells of the distal nephron, accompanied by mTORC1 activation. Kidney epithelial cells aberrantly proliferate in monolayer, resulting in a polycystic kidney-like morphology and lethal renal failure by 4 weeks of age. This phenotype is suppressed by rapamycin treatment, supporting the involvement of mTORC1 activation in the pathogenesis of BHDS [185, 186]. Flcn heterozygous knockout mice, mimicking affected BHDS patients, develop solid tumors, which demonstrate LOH of the remaining Flcn allele and similar histologies to human BHDS-associated tumors. mTOR activity, evaluated by western blotting, was high in these solid tumors [187]. On the other hand, another Flcn heterozygous mouse model showed suppressed mTORC1 activity in solid tumors and cysts which were evaluated by immunohistochemistry of phosphorylated S6 ribosomal protein on paraffin-embedded samples [188]. A third Flcn heterozygous mouse model exhibited increased phospho-S6 staining in large cysts and suppressed phospho-S6 staining in small cysts on paraffin-embedded samples [189]. In addition to these in vivo data, mTORC1 regulation by FLCN is shown to be cell type dependent [189–191].
A second FLCN-binding protein, FNIP2 (which is also known as FNIPL [192] or MAPO1 [193]), was identified by bioinformatics search [194]. FNIP2 is very similar to FNIP1 (identity, 49%; similarity, 74%) and shares the same characteristics as FNIP1 in binding to FLCN and AMPK. Hasumi et al. have shown that FNIP1 and FNIP2 can make hetero- or homomultimers, which can complex with FLCN and AMPK. This finding suggests that FLCN/FNIP1/FNIP2 may function as a tumor suppressor in a complex. Fnip1 homozygous knockout mice have B cell developmental defects and show no obvious phenotype in kidneys [195, 196]. Fnip2 homozygous knockout mice show no phenotype at all. However, kidney-targeted Fnip1 and Fnip2 double knockout mice exhibit completely identical phenotypes to kidney-targeted Flcn knockout mice [197]. This finding indicates that Fnip1 and Fnip2 may have redundant function and that FLCN and FNIP1/FNIP2 function coordinately as a tumor suppressor complex.
Other signaling pathways are also regulated by FLCN. Klomp et al. have compared gene expression profiles between BHDS-associated renal tumors and sporadic chromophobe RCC/oncocytoma and found that mitochondrial genes which are regulated by PPAR-γ coactivator 1α (PPARGC1A) were expressed significantly higher in BHDS-associated renal tumors [198]. Hasumi et al. have demonstrated that Flcn regulates Ppargc1a in vivo by analyzing muscle-targeted Flcn knockout mice. Flcn-deficient muscle shows increased mitochondrial biogenesis accompanied by increased Ppargc1a expression and a metabolic shift to oxidative phosphorylation, which is completely neutralized by the additional deletion of Ppargc1a [199]. It remains to be determined whether regulation of PPARGC1A activity by FLCN serves an essential role in FLCN tumor suppressor function. Hasumi et al. have shown suggestive data indicating that deletion of Ppargc1a in kidney-targeted Flcn knockout mice results in complete loss of hyperplastic cells, although aberrant kidney epithelial cell proliferation is seen in these animals and eventually causes lethal renal failure [199]. Flcn inactivation in murine cardiac muscle led to ATP overproduction, caused by aberrant mitochondrial biogenesis, AMPK suppression followed by mTORC1 activation, and cardiac hypertrophy, which was suppressed by rapamycin treatment or inactivation of Ppargc1a [200].
Recent evidence suggests that FLCN is a multifunctional protein. One of the important functions for FLCN is regulation of transcriptional activity of the basic-helix-loop-helix leucine zipper transcription factor, TFE3, a member of the microphthalmia-associated transcription factor (MiT) family. Under FLCN-deficient conditions, TFE3 translocates into the nucleus and has increased transcriptional activity [191, 201]. TFE3 regulation by FLCN might be essential for the role of FLCN as a tumor suppressor for the following reason. There is a rare subset of sporadic RCC, Xp11.2 translocation RCC, with translocations between TFE3 at Xp11.2 and a variety of genes, including ASPL, PRCC, NonO, PSF, and CLTC [202–204]. All of the proteins encoded by these TFE3 fusion genes maintain the C-terminal half of TFE3, where the basic-helix-loop-helix leucine zipper domain is located, and show nuclear TFE3 immunostaining in the corresponding Xp11.2 translocation RCC [205], suggesting that TFE3 constitutive activation leads to RCC development.
Moreover, FLCN is involved in the TGF-β signaling pathway [206, 207], ciliogenesis [208], and autophagy [209, 210]. The pathogenesis of lung cysts in BHDS has been uncertain for a long time. Identification of a FLCN-binding protein, plakophilin-4 (p0071), shed light on the molecular role of FLCN in cell-cell adhesion and cell polarity, which might be involved in the lung manifestations of BHDS [190, 211, 212]. Rho A signaling, which is regulated through p0071, is disordered under FLCN-deficient conditions. FLCN regulates cell-cell adhesions, and defects in this process may cause lung cyst formation [190]. Goncharova et al. have developed lung-targeted Flcn knockout mice and showed increased apoptosis in lung epithelium, which was caused by a dysregulated E-cadherin-LKB1-AMPK axis [213]. The multifunctionality of FLCN might explain the broad phenotype seen in Flcn knockout mice as well as the distinct manifestations of BHDS.
2.6.4 BHDS Research: Bench to Bedside
Currently there is no approved targeted therapy for BHDS. Part of the reason for this may be the rarity of BHDS and indolent nature of most BHDS-associated RCC. Based on kidney-targeted Flcn knockout mouse model results [185], mTORC1 inhibition might be a promising targeted strategy. In fact, Nakamura et al. treated advanced BHDS-related RCC with the mTORC1 inhibitor, everolimus, as a sixth-line therapy after disease was refractory to IL-2 (3 month, progressive disease (PD)), IFNα (3 month, PD), S-1(28 month, PD), sorafenib (1 month, PD), and sunitinib (4 month, PD). Even though everolimus was used as a sixth-line systemic therapy, it displayed a relatively long-term effect (SD for 7 month). Further progress in both basic research and translational research will be necessary for developing successful treatments for advanced RCC in BHDS.
2.7 Tuberous Sclerosis Complex (TSC)
Tuberous sclerosis complex (TSC) is an autosomal dominant hereditary hamartoma syndrome, which is caused by germline loss of function mutations in TSC1 or TSC2 genes. Disease manifestations are seen in multiple organs, including the skin, brain, heart, lung, eye, and kidney, with widely variable clinical presentations even among relatives (Fig. 2.6a–h) [214, 215]. Affected individuals are highly predisposed to develop renal angiomyolipomas, which are benign tumors in most cases. It should also be noted that TSC patients can develop renal epithelioid angiomyolipomas with malignant potential and, in rare cases, RCC with a characteristic histology. Since epithelioid angiomyolipoma is sometimes misdiagnosed for RCC, it is important to correctly distinguish these renal lesions in TSC patients.
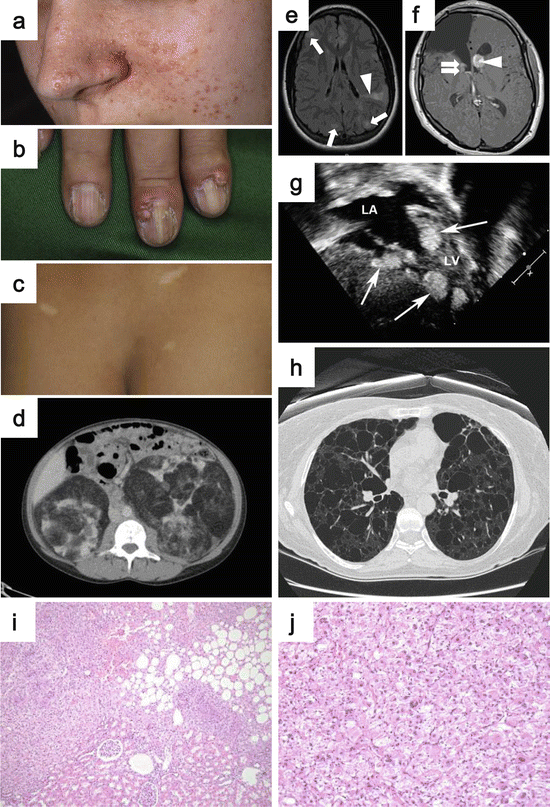
Fig. 2.6
Clinical manifestations of TSC. (a) Angiofibromas on the centrofacial area. (b) Ungual fibromas arising from the nail bed. (c) Hypomelanotic macules are observed frequently in TSC patients. (d) CT image showing bilateral multifocal angiomyolipomas. (e) MRI image demonstrating cortical dysplasia, which is observed very frequently in TSC patients (arrows: tubers, arrowhead: radial migration line). (f) MRI image indicating subependymal nodules (SEN) with arrows and subependymal giant cell astrocytoma (SEGA) with arrowhead. (g) Echocardiogram of cardiac rhabdomyomas. (h) Chest CT image demonstrating lymphangioleiomyomatosis (LAM). (i) Histology of angiomyolipoma. (j) Histology of epithelioid angiomyolipoma composed of pleomorphic cells with large hyperchromatic nuclei and abundant eosinophilic cytoplasm (Images from Northrup et al. [215] (a–h), and Kato et al. (i, j) [254])
2.7.1 Clinical Manifestations of TSC
TSC has been underdiagnosed because of the variable severity of manifestations among affected individuals [216]. Through the discoveries of the causative genes and establishment of diagnostic criteria, significant advancements have been made in the management of TSC. Currently its prevalence is estimated at 1/6000 to 1/10,000 of live births [215, 217]. The second International TSC Consensus Conference was held in 2012 and revised the clinical diagnostic criteria published in 1998. The identification of a pathogenic mutation in TSC1 or TSC2 in genomic DNA is sufficient for a definitive diagnosis of TSC. Since conventional genetic testing does not identify germline mutations in TSC1/2 in a significant population (10–25%) of TSC patients, a negative outcome of a genetic test does not exclude TSC. Here, an outline of TSC manifestations will be described. For details of the clinical diagnostic criteria, the reader is referred to the literature [216].
2.7.1.1 Extrarenal Manifestations of TSC
Dermatologic features are seen in almost 100% of TSC-affected individuals, which can be easily recognized by physical examination (Fig. 2.6a–c). The prominent manifestations are skin hamartomas, which include angiofibromas, fibrous cephalic plaques, ungual fibromas, and shagreen patches. Facial angiofibromas are seen in 75% to 93% of affected individuals [218–220]. Angiofibromas are red to pink papules with smooth surface, which distribute over the centrofacial area (Fig. 2.6a). Histologically, dermal fibrosis, coarse collagen bundles, stellate fibroblasts in the upper dermis, and capillary dilation are seen with atrophic sebaceous glands [221, 222]. Second-hit somatic TSC1 or TCS2 mutations were identified in cultured fibroblasts isolated from angiofibromas of TSC patients, supporting the idea that UV-induced DNA damage caused second-hit mutations in skin fibroblasts resulting in hamartoma formation [223]. There are two reports, suggesting a possible phenotypic overlap of skin hamartoma between TSC and Birt-Hogg-Dubé syndrome (BHDS). One publication reports angiofibromas in BHDS, while the other reports fibrofolliculomas in TSC [222, 224]. Clinicians should be aware that these overlapping clinical manifestations can sometimes make the differential diagnosis of TSC and BHDS challenging. In addition, angiofibromas are also seen frequently in another hereditary neoplastic syndrome, multiple endocrine neoplasia type 1 (MEN1), which do not develop kidney neoplasia [225]. Fibrous cephalic plaques (forehead fibrous plaques) are seen in around 25 to 46% of TSC-affected individuals [215, 220]. Fibrous cephalic plaques are histologically similar to angiofibromas, with remarkably sclerotic collagen tissue [221]. Ungual fibromas show later onset and are seen in 20 to 80% of patients in an age-dependent manner (Fig. 2.6b) [226, 227]. They are skin-colored or red nodules, arising from the nail bed of fingers or toes. Histologically they are similar to angiofibromas or fibrous cephalic plaques [215, 218, 220, 228]. Another proliferative skin manifestation is the shagreen patch, which is specific for TSC and seen in 50 to 80% of TSC-affected individuals in their first decade of life [219, 220, 227¸ 228]. They usually appear as large plaques on the lower back with the rough surface resembling an orange peel. Histologically it is a connective tissue hamartoma composed of vascular structures, adipose tissue, collagen, smooth muscle, and cutaneous appendages [229]. Another set of skin manifestations are large hypomelanotic macules and tiny confetti-like macules. Hypomelanotic macules are observed frequently in 65 to 90% of TSC patients (Fig. 2.6c) [218, 220, 227]. Confetti-like macules are numerous scattered tiny white hypomelanotic macules usually covering the arms and legs, which are seen in about 50% of affected patients [218, 226].
Central nervous system features are also very common in TSC. Cortical dysplasia, including cortical tuber and cerebral white matter radial migration lines, which can be diagnosed by MRI, are observed in 90% of patients (Fig. 2.6e). Cortical dysplasia is associated with intractable epilepsy and learning difficulties in TSC [215]. Subependymal nodules (SEN) and subependymal giant cell astrocytomas (SEGA) are observed in 80% of TSC patients (Fig. 2.6f) [215]. They are basically benign and slow growing, but can cause serious neurological morbidity. Cardiac rhabdomyomas can occur in 50% of cases (Fig. 2.6g) [218–220], which are rarely observed in non-TSC patients.
Lymphangioleiomyomatosis (LAM) is one of the major manifestations of TSC. Histologically, benign-appearing smooth muscle cells (LAM cell) are infiltrating into lymphatics, airways, blood vessels, and alveolar septa, and thin-walled lung cystic changes, which are the cause of destruction of alveolar structures, are observed [230, 231]. Upon high-resolution CT scanning, at least 30 to 40% of TSC-affected females present cystic pulmonary parenchymal changes, which are consistent with LAM (Fig. 2.6h) [232, 233]. The cystic changes of lung, consistent with LAM, are observed in about 10% of male TSC individuals, but symptomatic LAM in males is rare [234]. The risk of LAM is age dependent, increasing by 8% each year. The prevalence of LAM in females reaches 80% by 40 years of age [233]. Cudzilo et al. have reported that 12.5% of TSC patients with LAM eventually die from LAM. The origin of LAM cells is unknown. Ninety-three percent of TSC patients with LAM have concurrent renal angiomyolipomas, and 100% have uterine PEComas (tumors showing perivascular epithelioid cell differentiation) [235], suggesting that these extra lung manifestations might be the source of LAM cells.
2.7.1.2 Kidney Manifestations of TSC
Angiomyolipoma is the major kidney manifestation of TSC, which can cause the most severe clinical symptoms. Angiomyolipomas are frequently seen bilaterally and multifocally in kidneys of nearly 80% of TSC-affected individuals (Fig. 2.6d) [236]. Angiomyolipomas can also develop in other organs including the liver [237]. Renal angiomyolipoma is a benign mesenchymal clonal neoplasm composed of variable proportions of hyalinized thick-walled dysmorphic blood vessels, immature spindle-shaped smooth muscle-like cells, and mature adipose tissue (Fig. 2.6i) [238, 239]. Karbowniczek et al. have microdissected each component of sporadic angiomyolipomas and demonstrated that all three components have LOH at the TSC2 locus and shown that all cell components have immunoreactivity to anti-phospho-S6 antibody, supporting mTORC1 activation presumably caused by loss of TSC2 [240]. This may support the idea that all the components of angiomyolipomas are derived from a common progenitor cell [241]. It has been postulated that the origin of angiomyolipoma is a renal mesenchymal precursor cell or a neural crest lineage cell [242, 243]. Most renal angiomyolipomas behave biologically as a benign lesion and show favorable prognosis, although there have been reports of nodal involvement and extensions into the renal vein and inferior vena cava [244, 245]. On the other hand, angiomyolipomas confer a risk to TSC-affected patients by causing chronic kidney disease (CKD) [246] and hemorrhage [247]. The abnormal vascular components of larger angiomyolipomas have a tendency to develop aneurysms, which can rupture and cause patients to go into shock [248, 249].
Angiomyolipomas contain a subset of the smooth muscle-like cells, which appear epithelioid with clear to pale eosinophilic granular cytoplasm and focally associate with the blood vessels. This distinctive cell type is called a perivascular epithelioid cell or PEC, which is shared among a family of mesenchymal neoplasms known as “PEComas” (tumors showing perivascular epithelioid cell differentiation). PEComas include angiomyolipomas, lymphangiomyomatoses, and clear cell “sugar” tumor of the lung [250]. As mentioned above, angiomyolipomas can display extremely variable proportions of each component. Angiomyolipomas, which display dominantly or exclusively epithelioid cells, are classified as epithelioid angiomyolipomas or epithelioid PEComas (Fig. 2.6j) [235]. It is important to note that epithelioid angiomyolipomas could be misdiagnosed as RCC. Epithelioid angiomyolipoma is histologically characterized by polygonal cells with eosinophilic to clear cytoplasm, prominent nucleoli, occasional marked nuclear atypia, and pleomorphic forms (Fig. 2.6j), forming solid arrangements [235, 251–254]. More importantly, epithelioid angiomyolipomas can be malignant neoplasms, which metastasize and cause death, especially in cases that show malignant histology [252, 253, 255, 256]. In rare cases, typical angiomyolipomas can become malignant with epithelioid or sarcomatous transformation [257, 258]. It is important to consider the possibility of epithelioid angiomyolipoma when a high-grade epithelioid renal neoplasm is observed in a TSC patient or is found coexisting with a conventional AML [254]. Immunohistochemistry is extremely useful for differential diagnosis of epithelioid angiomyolipoma. PEC are positive for melanocytic antigens (HMB-45 and melan-A) as well as smooth muscle-specific actin and negative for epithelial markers, EMA, and cytokeratin [254].
The incidence of RCC in TSC-affected individuals is thought to be very rare and estimated to be 2 to 3%, which is comparable to the incidence of sporadic RCC in the general population [259, 260]. There have been many case reports of TSC-associated RCC with a variety of histologies. But there has not been any systematic evaluation and/or classification of these TSC-associated RCC. Recently, two groups have evaluated and classified TSC-associated RCC independently [261, 262]. Both groups have concluded that the RCCs in TSC show distinct histology and character, which differ from sporadic RCC in non-TSC general populations. Guo et al. have analyzed 57 RCCs from 18 TSC-affected patients. They describe unique clinicopathologic features of TSC-associated RCC including female predominance, younger age at diagnosis, multiplicity, association with angiomyolipoma, favorable clinical course, and three distinct histologic patterns as follows: (1) carcinoma resembling renal angiomyoadenomatous tumors (RAT-like) or RCC with smooth muscle stroma (30%), (2) carcinoma resembling sporadic chromophobe-type RCC (chromophobe-like) (59%), and (3) a unique granular eosinophilic-macrocystic histology (11%) [261]. Yang et al., have analyzed 46 RCC from 19 TSC patients and classified them into three categories based on morphologic, immunologic, and molecular profiles as follows: (1) “TSC-associated papillary RCC” with prominent papillary architecture and loss of SDHB expression (52%), (2) hybrid oncocytic/chromophobe tumor (HOCT) (33%), and (3) unclassified (15%) [262]. In both studies, HMB-45 negativity and Pax8 positivity were tested to exclude epithelioid angiomyolipoma. Both studies share distinct clinicopathologic characteristics of TSC-associated RCC.
2.7.2 Genetics of TSC
Through linkage analysis of TSC families, causative germline mutations in TSC1 and TSC genes were identified [263–265]. TSC1 localizes on chromosome 9q34, encoding an 1164 amino acid 140kD protein, hamartin. TSC2 localizes on chromosome 16p13, encoding an 1807 amino acid 200kD protein, tuberin. Seventy-five to 90% of TSC patients diagnosed through clinical criteria exhibit pathogenic germline mutations in either TSC1 or TSC2. Extensive genetic analysis of the TSC1 and TSC2 genes in TSC patients have identified a broad spectrum of mutations [219, 266–269]. To date, more than 500 unique TSC1 sequence variants and 1400 unique TSC2 sequence variants, which do not include nonpathogenic variants, have been reported (http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC1, http://chromium.lovd.nl/LOVD2/TSC/home.php?select_db=TSC2 ). Missense mutations, large genomic deletions, and in-frame deletions are very rare in TSC1. The germline mutation frequency in TSC2 is higher than TSC1. Especially in de novo cases, mutation frequency in TSC2 is reported to be two to ten times higher than in TSC1 [219, 268–273]. On the other hand, the mutation frequency in TSC pedigrees which segregate across multiple generations is approximately equal in TSC1 and TSC2 [180]. This might be explained by the fact that TSC1 mutations are associated with a less severe phenotype in TSC patients [219, 268]. LOH in TSC1 or TSC2 is consistently observed in most TSC-associated neoplastic lesions including angiomyolipomas, but rarely observed in cerebral cortical tubers [274, 275]. This indicates that TSC1 and TSC2 are classical tumor suppressor genes which follow the Knudson two-hit theory [276]. Although TSC is an autosomal hereditary syndrome, the sporadic cases, which have acquired de novo mutations without family history, are predominant. It has been estimated that about 66% to 83% of all TSC patients are sporadic cases [219, 268, 273, 277]. Therefore, although TSC is a hereditary syndrome, one should notice that lack of family history does not exclude TSC from the differential diagnosis.
2.7.3 Molecular Function of TSC1/TSC2
Both TSC1 and TSC2 are confirmed to function as tumor suppressor genes by in vitro and in vivo experiments [278–280]. TSC1 encodes a 140kD protein, TSC1 (hamartin), which does not have any known functional domains. TSC2 encodes a 200kD protein, TSC2 (tuberin), which has a GAP (GTPase-activating protein) domain in its c-terminal region. TSC1 and TSC2 share no homology and form a heterodimer [281, 282] to function as a GAP toward the small G-protein Rheb (Ras homolog enriched in the brain). As expected from the fact that both mutations in TSC1 and TSC2 cause a single disease, TSC1 and TSC2 function as a complex. TSC1 binds to TSC2 and stabilizes it by preventing ubiquitin-mediated degradation [283, 284]. The GAP activity is essential for TSC1/TSC2 tumor suppressor function [285]. Indeed, missense germline mutations are frequently found in TSC patients in the GAP coding regions of TSC2, underscoring the importance of GAP activity for TSC2 tumor suppressor function [286]. The TSC1/TSC2 complex activates Rheb GTPase and accelerates the conversion of GTP-bound Rheb to GDP-bound Rheb, resulting in inhibition of mTORC1 (composed of mTOR, RAPTOR, mLST8, and PRAS40) activity [287–289]. The TSC1/TSC2 complex receives upstream signals from many canonical signaling molecules including AKT, AMPK, Ras-ERK-RSK, Wnt-GSK3β, and HIF1α-REDD1 and works as a central hub of signaling transduction, which regulates mTORC1 activity [290]. Inactivation of TSC1 or TSC2 causes aberrant accumulation of GTP-bound Rheb resulting in constitutive activation of mTORC1 [291]. mTORC1 has a pivotal role in regulation of cell growth and proliferation and is activated in a majority of cancers [292].
Therapies that target mTORC1 using rapalogues have shown a very dramatic effect on angiomyolipoma and LAM in TSC patients. The problem is that the mTORC1 effect is cytostatic and termination of rapalogue treatment causes regrowth of tumors [293]. Although there were two advanced cases reported that did not respond to rapalogue treatment [294, 295], there are several case reports of advanced epithelioid angiomyolipomas treated with rapalogues with dramatic responses [296–298]. One thing to be considered is that constitutive activation of mTORC1 by loss of TSC1/2 function suppresses insulin signaling-mediated PI3K/AKT activation through a feedback loop [299]. So mTORC1 inhibition by rapalogues might release this feedback loop and reactivate PI3K/AKT signaling.
2.8 Cowden Syndrome (CS)/PTEN Hamartoma Tumor Syndrome (PHTS)
Cowden syndrome (CS)/PTEN hamartoma tumor syndrome (PHTS) is an autosomal dominant hereditary cancer syndrome, which is caused by germline mutations in a tumor suppressor gene PTEN. CS/PHTS predisposes patients to develop breast, thyroid, kidney, uterine, and other types of cancers as well as benign neoplasia and neurodevelopmental disorders. Because of its rareness and difficulty to diagnose due to the wide spectrum of manifestations, CS tends to be underestimated as a cause of kidney cancer. PTEN hamartoma tumor syndrome (PHTS) was defined to describe patients having germline mutations in PTEN [300]. In this chapter the term CS will be used to represent CS/PHTS.
2.8.1 Clinical Manifestations of CS/PHTS
CS was first reported in 1962 describing a case with a family history and was named after the first patient’s name [301]. This rare syndrome is inherited in an autosomal dominant manner with an estimated prevalence of at least 1 in 200,000 individuals [302].
CS displays a wide range of clinical characteristics including benign neoplasia, malignancies, central nervous system anomalies, and dysmorphic characteristics [303]. Mucocutaneous manifestations are the most common manifestations of CS, which include trichilemmomas (hair follicle hamartoma), papillomatous papules, and acral/plantar keratoses, and are present in 99% of CS patients by their third decade of life (Fig. 2.7b, c) [304]. Other commonly observed features seen in CS patients are macrocephaly (Fig. 2.7a), dolicocephaly, and dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos) [304]. In addition, affected patients can develop benign tumors that include colorectal polyposis, thyroid goiter/nodules, lipomas, fibromas, and proliferative breast changes [303].
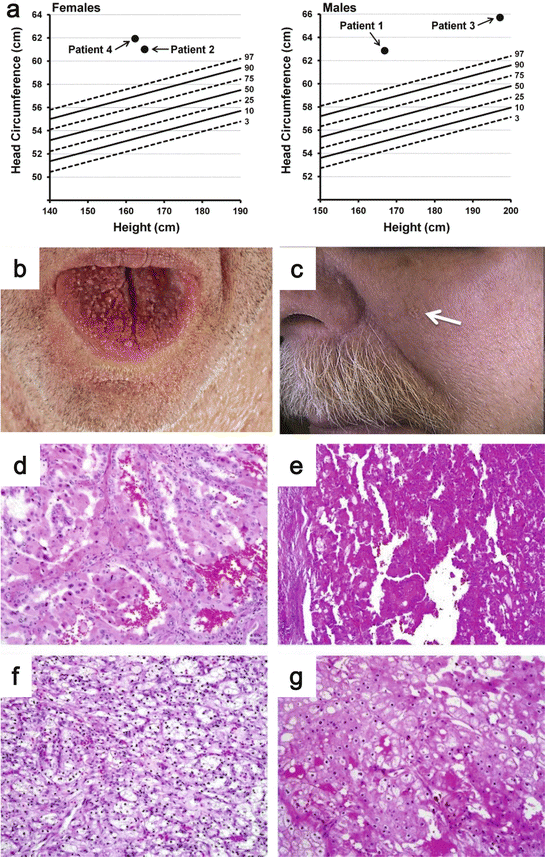
Fig. 2.7
Clinical manifestations of CS. (a) Macrocephaly is commonly observed in CS patients. (b) Mucocutaneous manifestations are the most common in CS patients. Image shows papillomatous papules on dorsum of the tongue. (c) Cutaneous verrucous papule over the centrofacial area. (d–g) Renal tumor histology in CS patients showing papillary type 1 RCC (d, e), ccRCC (f), and chromophobe RCC (g) (Images from Shuch et al. [313])
Individuals affected with CS are at risk throughout their lifetime to develop a variety of cancers, which can be bilateral and multifocal, similar to other inherited cancer syndromes. Affected women have the lifetime risk for breast cancer ranging from 67% to 85% [305–307], which is even higher than the lifetime risk of hereditary breast and ovarian cancer (HBOC) syndrome caused by germline mutations of BRCA1 or BRCA2 [308]. CS patients can have a variety of benign breast lesions, which are difficult to differentiate from cancers [309]. Careful and close follow-up of breast lesions is required. The lifetime risk for thyroid cancer is from 7.8% to 38% [305–307]. Among the thyroid cancers, the papillary type is the most common histology (52%), followed by a follicular variant of papillary (28%) and follicular (14%) [310]. Since most CS patients have multinodular thyroids, goiter (73%), and Hashimoto’s disease (27%), careful differential diagnosis and close follow-up are also necessary [310]. Affected women have an increased risk of endometrial cancer with a lifetime risk of 21%–28% [305, 307]. The lifetime risk for colorectal cancer is 9%, while 93% of affected patients who had a GI tract endoscopy were found to have polyps [311].
Mester et al. have analyzed the prevalence and histology of RCC among 219 CS patients who were confirmed to have pathogenic germline mutations in PTEN [312]. Nine of the 219 patients had a medical history of RCC, which means the age-adjusted standardized incidence ratio (SIR) is 31.7. Differently from sporadic RCC, the SIR is higher for females (46.7) than males (21.6). The lifetime risk of RCC for CS affected patients is calculated as 34% [305]. Shuch et al. have reported a higher incidence of RCC cases among the CS patients (4 in 24 patients) and have pointed out that RCC is an underappreciated feature of CS [313]. A wide variety of histologies have been reported in CS-associated RCC (Fig. 2.7d–g). Mester et al. have reported that 75% of cases are papillary RCC and 25% of cases are chromophobe RCC [312]. Shuch et al. have reported 50% papillary RCC (Fig. 2.7d, e), 25% clear cell RCC (Fig. 2.7f), and 25% chromophobe RCC in CS (Fig. 2.7g) [313]. Clearly CS-associated RCCs have different characteristics from other types of hereditary kidney cancers. Further analysis with a larger cohort will be required to define the histological spectrum of CS-related RCC. Although it has to be confirmed in a larger cohort, CS-associated RCC seems to be less malignant. To date there are no reports of metastatic RCC in CS [303, 313].
Another characteristic of RCC in CS is the absence of family history of RCC, although the total number of reported CS-associated RCCs is limited [303, 313]. Shuch et al. discuss that this is probably because of low disease penetrance and a high rate of de novo germline mutations in PTEN, which is estimated to be between 10.7% and 47.6% [313, 314]. Therefore, lack of RCC family history does not exclude a diagnosis of CS in a patient. Recognition of pathognomonic characteristics like mucocutaneous lesions, medical history of other type of cancers, GI hamartomas, and neurodevelopmental disorders would be important for clinicians to diagnose CS patients with RCC.
2.8.2 Genetics of CS/PHTS
Genetic linkage analysis of 12 CS families identified a responsible genetic locus on chromosome 10q22–23 in 1996 [315]. PTEN, a candidate tumor suppressor gene located on 10q23, was found to be mutated in cell lines of glioblastomas, prostate cancers, and breast cancers as well as in primary glioblastomas and other cancers [316, 317]. Subsequently, loss of function mutations of PTEN were found in the germline of CS kindreds [318–320]. The germline PTEN mutation spectrum includes all types of mutations located throughout the gene. Although the physiological meaning is unknown, there are significant correlations between promoter mutations and breast cancer incidence and between nonsense mutations and colorectal cancers [305]. There is no clear correlation between germline mutations in PTEN and a specific histology of RCC in CS [313]. LOH of PTEN has been analyzed in five cases of CS-associated RCC and was found in four cases, indicating that LOH might be the major mechanism for second-hit PTEN alterations driving RCC development in CS [313]. Mester et al. have reported negative PTEN immunohistochemistry staining in all 5 cases of analyzed CS-associated RCC. Negative PTEN staining might be a useful marker to suggest the possibility of CS-associated RCC, because PTEN expression is mostly positive in sporadic RCC [312]. Kondo et al. have reported that 5 of 68 (7.5%) cases of sporadic RCC exhibit somatic loss of function mutations and 25% of cases show LOH of PTEN, including 3 of the cases with somatic mutations in PTEN. Among the five somatic PTEN mutation cases, four cases were high-grade advanced ccRCC with poor prognosis. The other case was low-grade papillary RCC [321]. The biological behavior of CS-associated RCC and PTEN-inactivated sporadic RCC appears to be different. Recent exome sequencing studies have identified PTEN loss of function mutations in sporadic ccRCC, papillary RCC, and chromophobe RCC [50, 51, 171, 322].
2.8.3 Molecular Function of PTEN
PTEN is a 403 amino acid multifunctional protein, which has phosphatase activity both on lipid and protein [323–326]. The main tumor suppressor function of PTEN is maintaining the homeostasis of the phosphatidylinositol 3 kinase (PI3K)/AKT cascade [327–329]. In response to extracellular signaling, receptor tyrosine kinases, G-protein-coupled receptors, and RAS can activate PI3K, which converts phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3) [330]. Increased local PIP3 recruits many signaling molecules, including phosphatidylinositol-dependent kinase 1(PDK1) and AKT together, to the plasma membrane, where AKT is activated by PDK1 [331]. Activated AKT regulates many downstream biological effects, including proliferation, survival, cell polarity, motility, cell cycle, metabolism, and angiogenesis [332]. PTEN dephosphorylates PIP3 to PIP2, resulting in reduced AKT activity and antagonizes PI3K/AKT signaling pathways. One of the most important signaling molecules downstream of PI3K/AKT is mTOR. AKT activates mTORC1 (composed of mTOR, RAPTOR, mLST8, PRAS40) by phosphorylating TSC2 [333, 334] and PRAS40 [335], causing phosphorylation of p70 ribosomal protein S6 kinase and 4EBP1 to promote protein translation. mTORC1 regulates many cellular processes, including protein synthesis, lipid synthesis, autophagy, cell cycle, growth, and metabolism [336, 337]. Among them, the PI3K/AKT/mTOR/HIF1α axis has an important role in cancer development by regulating glucose metabolism as well as angiogenesis [338, 339]. Apart from its tumor suppressor role in the PI3K/AKT axis, PTEN has a phosphatase independent role in the nucleus to regulate chromosomal stability, double-strand DNA break repair, and the cell cycle [327, 340, 341]. These findings suggest that targeting the PI3K/AKT/mTOR axis itself may not be sufficient to treat PTEN-deficient cancers. Targeting the loss of function effect of PTEN in the nucleus might be useful in combinatorial therapy or next-generation targeted therapy for PTEN-deficient cancers.
2.9 BAP1 Germline Mutations (BAP1 Cancer Syndrome and BAP1 Tumor Predisposition Syndrome)
BAP1 (BRCA1-associated protein 1) is a tumor suppressor gene [342, 343] which resides on chromosome 3p21 and is frequently deleted in ccRCC. Recently a novel autosomal dominant tumor predisposition syndrome, associated with loss of function germline mutations in BAP1, has been proposed. [344, 345] BAP1 germline mutations predispose patients in a familial setting to develop a variety of tumors including ccRCC (Fig. 2.8a) [344, 346, 347]. BAP1 inactivation also contributes to the development and progression of sporadic ccRCC, which underscores the importance of gaining a better understanding of this emerging cancer syndrome [50–54].
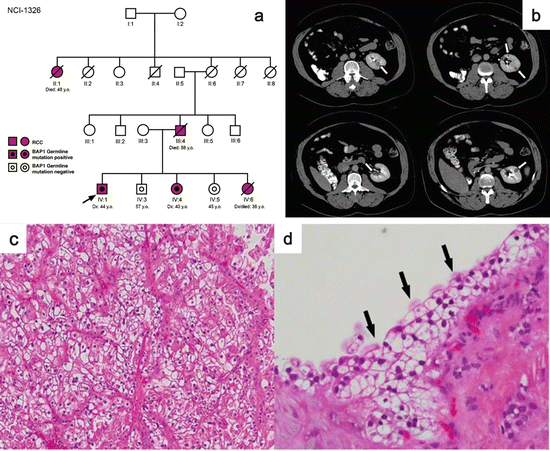
Fig. 2.8
Clinical manifestations of BAP1 tumor predisposition syndrome. (a) Pedigree of BAP1 tumor predisposition syndrome family. Red symbols indicate individuals with RCC. (b) CT image of affected individual following right radical nephrectomy demonstrating multifocal left renal lesions. (c) Histology of solid ccRCC in affected individual. (d) Histology of atypical renal cyst with clear cell lining (Images from Farley et al. [347])
2.9.1 Clinical Manifestations of BAP1 Tumor Predisposition Syndrome
BAP1 germline mutations predispose patients to develop malignant mesothelioma, uveal melanoma, cutaneous melanoma, and new category of tumor “melanocytic BAP1-mutated atypical intradermal tumors” (MBAITs) [344]. MBAIT is a newly proposed term to describe atypical melanocytic tumors that were previously diagnosed using various terminologies [348–351]. Carbone et al. have performed meta-analysis of published families with BAP1 germline mutations [348–351] and have shown that MBAITs are the most highly penetrant manifestation of the BAP1 cancer syndrome, seen in 66.7% of affected individuals. MBAITs are often associated with a compound nevus or intradermal nevus, grow very slowly, and are thought to be benign tumors. MBAITs are characterized histologically as intradermal lesions with large epithelioid and spindle-shaped melanocytes (MBAITs cells), which show cellular atypia and pleomorphic/hyperchromatic nuclei, but no mitotic figures or Ki67 staining. Through meta-analysis, Carbone et al. have reported the prevalence of other tumors in BAP1-mutated individuals as follows: malignant mesothelioma (MM, 21%), uveal melanoma (UM, 17.7%), and cutaneous melanoma (CM, 12.9%). None of these tumors has been observed in non-affected family members, suggesting that these manifestations are significant features of the BAP1 cancer syndrome [344]. Popova et al. have reported that among 6 of the 11 families with BAP1 cancer syndrome, 9 affected individuals presented with RCC [346]. Farley et al. also have reported a novel germline mutation in BAP1, which predisposes to familial ccRCC [347]. These findings strongly support RCC as a manifestation of the BAP1 cancer syndrome. To date, there is no report regarding the pathological analysis of BAP1 cancer syndrome-associated RCC and no consensus of histological features, which would be useful for diagnosis of BAP1 cancer syndrome-associated RCC. However, based on two reports, bilateral multifocal early-onset ccRCC with high Fuhrman grade might be characteristic of BAP1 cancer syndrome-associated RCC (Fig. 2.8b–d) [346, 347]. There are many reports suggesting the involvement of other types of cancers in BAP1 cancer syndrome, i.e., breast cancer, meningioma, lung cancer, neuroendocrine carcinoma, and basal cell carcinoma [350, 352–355]. To define the true tumor spectrum of BAP1 cancer syndrome, a large-scale recruitment of affected families and intensive analysis would be required.
2.9.2 Genetics of BAP1 Tumor Predisposition Syndrome
BAP1 inactivating somatic mutations were first identified by whole-exome sequencing of metastatic uveal melanomas, which had chromosome 3 monosomy [356]. Additional Sanger sequencing of all BAP1 exons revealed the frequent loss of function mutations of BAP1 in metastasizing uveal melanomas (26/31; 84%). Subsequently, somatic inactivation mutations were found in 23% of malignant pleural mesothelioma [357]. Following these findings, germline mutations in BAP1 were reported as predisposing to malignant mesothelioma, melanocytic tumors, uveal melanoma [348–352], and RCC [346, 347]. From 7.5 to 14% of cases of sporadic ccRCC are reported to have somatic inactivating mutations in BAP1, which underscores the significance of loss of BAP1 function in developing ccRCC [50–54]. Most of the germline mutations reported to date are nonsense or insertion/deletion mutations causing frameshift and premature terminations [358]. To date, there is no report describing distinct genotype-phenotype correlations in BAP1 cancer syndrome.
Related posts:
 Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
 Forthcoming Drugs for Metastatic Renal Cell Carcinoma Therapy
Forthcoming Drugs for Metastatic Renal Cell Carcinoma Therapy
 Immunotherapy for Renal Cell Cancer (RCC)
Immunotherapy for Renal Cell Cancer (RCC)
 Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
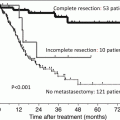 Treatment Overview
Treatment Overview
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


