High-grade serous
Low-grade serous
High-grade endometrioid
Low-grade endometrioid
Clear cell
Mucinous
Unspecified carcinoma
No.
1501
70
64
14
58
16
166
BRCA1 (%)
10.3
4.3
6.3
0
6.9
0
8.4
BRCA2 (%)
5.7
1.4
4.7
0
0
0
5.4
BRCA1/2 (%)
16.0
5.7
10.9
0
6.9
0
13.9
Table 2.2
Histological and molecular subtypes of epithelial ovarian cancer
High-grade serous | Low-grade serous | High-grade endometrioid | Low-grade endometrioid | Clear cell | Mucinous | |
|---|---|---|---|---|---|---|
Genomic alterations | TP53 BRCA1/2 Other HRR genes | BRAF KRAS PTEN PIK3CA | BRCA1/2 | PTEN PIK3CA CTNNB1 ARID1A BRAF | ARID1A PIK3CA | KRAS CDKN2A PIK3CA BRAF TP53 |
Copy number alterations | – | – | – | – | ERBB2 | ERBB2 |
Several founder mutations have been observed in the specific population, for example, the 187delAG and 5385insC mutations in BRCA1 and the 6174delT mutation in BRCA2 have been identified in the Ashkenazi Jewish population [30, 31]. In Japanese population, it was reported that the L63X and Q934X mutations in BRCA1 were the founder mutations with high frequency in hereditary ovarian cancer families [17], and it has been reported that the L63X is a founder mutation with the highest frequency in Japanese breast cancer families [32, 33].
The penetrance of BRCA1/2 gene mutation in ovarian cancer is lower than that in breast cancer. A lifetime risk for ovarian cancer in BRCA mutation carriers was estimated to be 8–62% in different populations; however, that for breast cancer was 41–90%. A meta-analysis of these published data showed the average cumulative risks for breast and ovarian cancer by age 70 years for BRCA1 mutation carriers were 57% and 40%, respectively. For BRCA2 mutation carriers, they were 49% and 18%, respectively, in the meta-analysis [5, 24, 34–42]. In a recent prospective study, the estimated average cumulative risks for breast and ovarian cancer by age 70 years for BRCA1 mutation carriers were 60% and 59%, respectively. In addition, for BRCA2 mutation carriers, they were 55% and 16.5%, respectively [39]. A subsequent alteration or silencing in the second copy of the gene without the hereditary mutation is believed to be necessary for the initiation of cancer development, so the risk of breast and ovarian cancer with BRCA1/2 mutations is various, even within families with the same mutation. In an international observational study of 19,581 carriers of BRCA1 mutations and 11,900 carriers of BRCA2 mutations in 33 countries on 6 continents, 12% of the BRCA1 mutation carriers and 6% of the BRCA2 mutation carriers were diagnosed with ovarian cancer, and 46% of the BRCA1 mutation carriers and 52% of the BRCA2 mutation carriers were diagnosed with breast cancer [43]. As described above, BRCA1/2 mutation carriers have a high risk for both breast cancer and an ovarian cancer, so there was a need to consider more intensive screening and prevention strategies such as chemoprevention and prophylactic surgery.
It has been reported that some pathological features are observed more frequently in breast and ovarian cancer patients with BRCA1/2 mutation. For example, breast cancers with BRCA1/2 mutation are characterized as ER/PR and HER2 negative: triple negative [44–49]. In ovarian cancers with BRCA1/2 mutation, high-grade serous carcinoma is a major histological subtype, although endometrioid and clear cell carcinomas also have been reported in the BRCA-related ovarian cancers [21, 25–27, 50–53]. Mucinous type is very rare in the population [25, 27]. In Japanese hereditary breast and ovarian cancer families, the major histological type of BRCA-associated ovarian cancers was serous carcinoma in 81% of tumors, and only one case was clearcell carcinoma. No tumor with mucinous carcinoma occurred in these families [17]. Mucinous carcinomas appear to be related to other gene mutations; KRAS and TP53 [54]. Borderline epithelial ovarian tumors are not associated with a BRCA1/2 mutation [21]. Although non-epithelial ovarian carcinomas are not significantly associated with a BRCA1/2 mutation, sex cord tumors may be associated with Peutz-Jeghers syndrome, and Sertoli-Leydig cell tumors are caused by germline mutations in the DICER1 gene [55–61].
Several studies have reported that BRCA mutation-positive women with ovarian cancer showed more favorable survival outcomes compared with mutation-negative women [62–67]. Figure 2.1 indicates that BRCA1/2 mutation carriers showed a more favorable survival than noncarriers (for BRCA1, HR = 0.78 [95% CI, 0.68–0.89], P < 0.001, and for BRCA2, HR = 0.61 [95% CI, 0.50–0.76], P < 0.001) in a pooled analysis from 26 observational studies that included invasive epithelial ovarian cancer cases from BRCA1/2 mutation carriers (n = 1213) and noncarriers (n = 2666) [63]. The 5-year overall survival was 36% for noncarriers, 44% for BRCA1 carriers, and 52% for BRCA2 carriers. In a population-based case-control study of women with invasive epithelial (non-mucinous) ovarian cancer (n = 1001), patients carrying germline mutations of BRCA1/2 had improved rates of progression-free survival (median, 20 months vs 16 months; not statistically significant) and overall survival (median, 62 months vs 55.5 months; P = 0.031) [62]. Survival outcomes appear to be most favorable for BRCA2 mutation carriers [63]. An observational study of 1915 women with ovarian cancer from the University of Washington (UW) gynecologic tissue bank and from the Gynecologic Oncology Group (GOG) phase III clinical trials (n = 1345) showed that patients with a BRCA2 mutation from the GOG trials had significantly longer progression-free survival (HR, 0.60; 95% CI, 0.45–0.79; P < 0.001) and OS (HR, 0.39; 95% CI, 0.25–0.60; P < 0.001), compared with those without mutations [22].
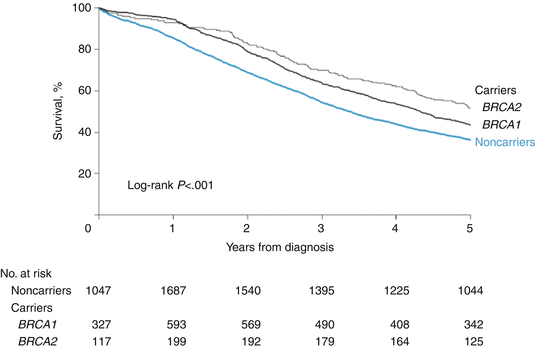
Fig. 2.1
Association between BRCA1/2 mutations and survival in women with invasive epithelial ovarian cancer. BRCA1 and BRCA2 mutation carriers showed a more favorable survival than noncarriers (for BRCA1, HR = 0.78 [95% CI, 0.68–0.89], P < 0.001, and for BRCA2, HR = 0.61 [95% CI, 0.50–0.76], P < 0.001) in a pooled analysis from 26 observational studies that included invasive epithelial ovarian cancer cases from BRCA1/2 mutation carriers (n = 1213) and noncarriers (n = 2666). Kaplan-Meier analysis was adjusted for year of diagnosis and study [63]
BRCA mutation carriers appeared to be more responsive to cytotoxic chemotherapy compared with noncarrier patients [68]. Several studies have shown a higher response rate to platinum regimens and longer treatment-free intervals between relapses in BRCA mutation carriers compared with noncarriers [62, 63, 66, 69–71]. These clinical features of BRCA-associated ovarian cancer are attributed to homologous recombination repair deficiency in the absence of BRCA1/2 function, which results in an impaired ability of tumor cells to repair platinum-induced double-strand breaks [66, 70, 72]. Thereby conferring increased chemosensitivity and increased sensitivity to poly (ADP-ribose) polymerase (PARP) enzyme inhibition and other DNA-damaging chemotherapeutic agents such as pegylated liposomal doxorubicin (PLD) [68].
2.2.2 Ovarian Cancer Screening for Surveillance
Ovarian cancer screening with transvaginal ultrasound and CA-125 has not been shown to be sufficiently sensitive or specific. So far, there is no evidence that these screening are appropriate methods of substituting for ovarian cancer risk-reducing surgery [73, 74]. In recent large randomized controlled trial, the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS), which assessed multimodality screening with ultrasound and CA-125 versus either ultrasound alone or no screening, showed that a significant mortality reduction was not observed after a median of 11 years of follow-up; however, a prespecified analysis of death from ovarian cancer of multimodality screening versus no screening with exclusion of prevalent cases showed significantly different death rates (P = 0.021) [75, 76]. In this trial, the cases with increased risk of familial ovarian cancer were included in exclusion criteria. The NCCN Guidelines recommend that ovarian cancer screening with transvaginal ultrasound and CA-125 may be considered starting at age 30–35 years by the doctor’s discretion for women who have not selected the risk-reducing surgery [13]. GOG-0199 is a two-arm, prospective, nonrandomized study for managing the risk of ovarian cancer in high-risk women. One arm is women who elected RRSO, and the other is those who chose the ROCA (risk of ovarian cancer algorithm) surveillance using transvaginal ultrasound and CA-125. This 5-year follow-up period ended in November 2011 and the data has been analyzed [77].
2.2.3 Risk-Reducing Salpingo-Oophorectomy (RRSO)
The risk for ovarian cancer in BRCA1/2 mutation carriers is generally considered to be lower than the risk for breast cancer. However, due to the absence of reliable methods of early detection and the poor prognosis associated with advanced ovarian cancer, RRSO after completion of childbearing has been recommended for BRCA1/2 mutation carriers. The NCCN Guidelines recommend RRSO for women with BRCA1/2 mutation, typically between ages 35 and 40 years for women with a BRCA1 mutation [13]. For women with a BRCA2 mutation who have undergone efforts to maximize their breast cancer prevention (i.e., bilateral mastectomy), it is reasonable to delay RRSO until between ages 40 and 45 years since ovarian cancer onset tends to be later in women with a BRCA2 mutation [78]. RRSO should only be considered upon completion of childbearing.
The effectiveness of RRSO in reducing the risk for ovarian cancer in BRCA1/2 mutation carriers has been reported in various studies. In a meta-analysis including ten studies, RRSO was associated with a statistically significant reduction in the risk of BRCA-associated ovarian or fallopian tube cancer (HR = 0.21; 95% CI = 0.12–0.39) [78]. In an international observational study of 5783 women with a BRCA1/2 mutation, risk-reducing oophorectomy was associated with an 80% reduction (HR, 0.20; 95% CI, 0.13–0.30) in the risk of ovarian, fallopian tube, or peritoneal cancer in BRCA1/2 carriers and a 77% reduction in all-cause mortality (HR, 0.23; 95% CI, 0.13–0.39) [78]. RRSO reduces mortality at all ages in BRCA1 mutation carriers; however, RRSO is not associated with reduced mortality in those at the ages of more than 61 in BRCA2 mutations carriers [78]. Furthermore, in prospective, multicenter cohort study of 2482 women with BRCA1/2 mutations, RRSO was associated with lower all-cause mortality (10% vs 3%; HR, 0.40 [95% CI, 0.26–0.61]), breast cancer-specific mortality (6% vs 2%; HR, 0.44 [95% CI, 0.26–0.76]), and ovarian cancer-specific mortality (3% vs 0.4%; HR, 0.21 [95% CI, 0.06–0.80]) [79]. We have to take care that 1–4.3% risk of a primary peritoneal cancer has remained after RRSO [80–84]. The ovarian cancer risk and management were shown in Table 2.3 [13].
Table 2.3
Ovarian cancer risk and management
Ovarian cancer risk | Management | |
|---|---|---|
BRCA1 | Increased risk of OC | Consider RRSO at 35–40 year |
BRCA2 | Increased risk of OC | Consider RRSO at 45–50 year |
MMR genes | Increased risk of OC | Consider RRSO and hysterectomy at completion of childbearing |
BRIP1 | Increased risk of OC | Consider RRSO at 45–50 year |
RAD51C | Increased risk of OC | Consider RRSO at 45–50 year |
RAD51D | Increased risk of OC | Consider RRSO at 45–50 year |
PALB2 | Insufficient evidence for OC risk | ― |
TP53 | No increased risk of OC | ― |
Many studies have reported that RRSO reduced the risk for breast cancer in BRCA1/2 mutation carriers [80, 81, 83, 85, 86]. In a meta-analysis of all reports of RRSO published between 1999 and 2007, RRSO was associated with a statistically significant reduction in risk of breast cancer in BRCA1/2 mutation carriers (HR = 0.49; 95% confidence interval [CI] = 0.37–0.65), BRCA1 mutation carriers (HR = 0.47; 95% CI = 0.35–0.64), and BRCA2 mutation carriers (HR = 0.47; 95% CI = 0.26–0.84) [80]. Results of a prospective cohort study suggest that RRSO may be associated with a greater reduction in breast cancer risk for BRCA2 mutation carriers compared with BRCA1 mutation carriers [87]. Reductions in breast cancer risk for BRCA1/2 mutation carriers following RRSO may be associated with decreased hormonal exposure due to resection of the ovaries. In an international case-control study of 1439 patients with breast cancer and 1866 matched controls derived from a registry of BRCA1/2 mutation carriers, the risk reduction was greater if the oophorectomy was performed before age 40 (OR = 0.36; 95% CI, 0.20–0.64 for BRCA1 carriers) than after age 40 (OR = 0.53; 95% CI, 0.30–0.91), and no significant reduction was found for women aged 51 years or older in breast cancer risk [86]. However, the hazard ratio for breast cancer-specific mortality in BRCA1/2 mutation carriers was 0.76 (95% CI, 0.32–1.78; P = 0.53) for women with estrogen receptor-positive breast cancer and 0.07 (95% CI, 0.01–0.51; P = 0.009) for women with estrogen receptor-negative breast cancer [88].
RRSO is an opportunity for occult gynecologic cancer detection in BRCA1/2 mutation carriers. In studies of women with a BRCA1/2 mutation who underwent RRSO, occult gynecologic carcinomas and ovarian, tubal, or peritoneal cancer were identified in 4.5–9% of cases, and tubal intraepithelial carcinoma (TIC) was detected in 5–8% of cases [84, 89–92]. The fimbriae or distal tube was reported to be the predominant site of origin for these early malignancies found in patients with BRCA1/2 mutations [89, 92, 93].
In a prospective cohort of 462 women with BRCA1/2 mutation carriers, short-term hormone replacement therapy (HRT) in women undergoing RRSO does not negate the protective effect of bilateral prophylactic oophorectomy on subsequent breast cancer risk in BRCA1/2 mutation carriers [94]. Moreover, results of a case-control study of BRCA1 mutation carriers showed no association between use of HRT and increased breast cancer risk in postmenopausal BRCA1 mutation carriers [95]. However, there is no randomized study of the issue, so the use of HRT in BRCA1/2 mutation carriers undergoing RRSO should be carried out carefully [96, 97].
Salpingectomy has been performed in premenopausal women, and there have been some evidence regarding the safety and feasibility of this procedure [98, 99]. However, there is limited data regarding its efficacy in reducing the risk for ovarian cancer [100, 101]. In addition, BRCA1/2 mutation carriers undergoing salpingectomy alone may not get the 50% reduction in breast cancer risk of BRCA1/2 carriers following oophorectomy. Hence, the salpingectomy alone has not been recommended as the standard risk-reducing surgery in BRCA1/2 mutation carriers at this time.
The NCCN Guidelines recommend RRSO protocol [102]: (1) Perform operative laparoscopy. (2) Survey upper abdomen, bowel surfaces, omentum, appendix (if present), and pelvic organs. (3) Biopsy any abnormal peritoneal findings. (4) Obtain pelvic washing for cytology. (5) Perform total BSO, removing 2 cm of proximal ovarian vasculature/IP ligament, all tube up to the cornua, and all peritoneum surrounding the ovaries and tubes, especially peritoneum underlying areas of adhesion between tube and/or ovary and the pelvic sidewall. (6) Engage in minimal instrument handling of the tubes and ovaries to avoid traumatic exfoliation of cells. (7) Both ovaries and tubes should be placed in an endobag for retrieval from the pelvis. (8) Both ovaries and tubes should be processed according to SEE-FIM protocol [103]. (9) If occult malignancy or STIC is identified, provide referral to gynecologic oncologist. (10) The prevention benefits of salpingectomy alone are not yet proven. If considered, the fallopian tube from the fimbria to its insertion into the uterus should be removed.
Japan Society of Gynecologic Oncology guidelines 2015 for the treatment of ovarian cancer described procedures for the examination and management of HBOC. In the guidelines, it was recommended that RRSO only be performed by a gynecologic oncologist who is a member of the Japan Society of Gynecologic Oncology in cooperation with a clinical geneticist at a medical facility with an established genetic counseling system and cooperative pathologists, after review and approval by the institutional ethics committee [104]. In addition, the Gynecologic Oncology Committee of Japan Society of Obstetrics and Gynecology have proposed the requirement of RRSO for BRCA1/2 mutation carriers in more detail [105].
2.2.4 Chemoprevention
As regards the effect of oral contraceptives (OC) in BRCA1/2 mutation carriers, two meta-analyses showed significant reduction of the risk for ovarian cancer. In analysis of BRCA1/2 mutation carriers with (n = 1503) and without (n = 6315) ovarian cancer, OC use significantly reduced the risk for ovarian cancer by approximately 50% for both the BRCA1 mutation carriers (RR, 0.51; 95% CI, 0.40–0.65) and BRCA2 mutation carriers (RR, 0.52; 95% CI, 0.31–0.87) [106]. The other including one cohort study (N = 3181) and three case-control studies (1096 cases and 2878 controls) also showed an inverse association between OC use and ovarian cancer (OR, 0.58; 95% CI, 0.46–0.73), and the risks appeared to decrease with longer duration of oral contraceptive use [107]. Two meta-analyses showed that OC use is not significantly associated with breast cancer risk in BRCA1/2 mutation carriers [106, 108]. However, case-control studies in the analyses on the effect of OC use on breast cancer risk in BRCA1/2 mutation carriers have showed conflicting results.
2.3 Genes Other than BRCA1/2 Involved in Hereditary Ovarian Cancer
2.3.1 Mismatch Repair Genes (Lynch Syndrome)
Ovarian cancer is a component tumor of Lynch syndrome that is associated with germline mutations in mismatch repair genes (MLH1, MSH2, MSH6, MLH3, and PMS2) [109]. Lynch syndrome, also known as hereditary nonpolyposis colon cancer (HNPCC), accounts for 10–15% of all hereditary ovarian cancers [109] and is at increased risk for endometrial and ovarian cancers: up to 60% and 24%, respectively [110–113]. The loss of function of one of the mismatch repair proteins results in the accumulation of repeated nucleotide sequences phenotypically expressed as microsatellite instability (MSI). Several oncogenes and tumor suppressor genes contain microsatellites; impairment of MMR could cause mutations in many genes implicated in ovarian tumorigenesis [114–118]. BRCA-related ovarian cancers are associated with non-mucinous tumors; on the other hand, Lynch syndrome-associated ovarian cancers appear to be associated with both non-mucinous and mucinous tumors. Ovarian cancers in Lynch syndrome are mostly endometrioid or clear cell [119–123]. The cumulative lifetime risk of ovarian cancer is estimated to be 6–10% in MSH2 and MLH1 mutation carriers. An average age of diagnosis was 51 years in families associated with MLH1 mutations and 45 years in families associated with MSH2 mutations [113, 124, 125]. Lynch syndrome-associated ovarian cancers were more likely at diagnosis to be of low grade and early stage and generally showed a better prognosis [124, 126, 127]. Total abdominal hysterectomy and/or bilateral salpingo-oophorectomy are options that may be considered for risk reduction in women with mutation of mismatch repair genes who have completed childbearing [128–132]. No evidence has been showed to support routine transvaginal ultrasound and CA-125 testing in these mutation carriers because they have not been shown to be sufficiently sensitive or specific [128, 133–137].
2.3.2 Homologous Recombination Deficiency (HRD)-Related Genes
Homologous recombination (HR) plays in a repair of double-strand breaks (DSBs) [29]. A lot of proteins involved in homologous recombination are recognized to also contribute to hereditary cancer risk, e.g., BRCA1/2, ATM, PALB2, RAD51C, RAD51D, CHEK2, BARD1, Mre11, RAD50, NBS1, BRIP1, and Fanconi anemia proteins [3]. These proteins interact with BRCA1/2 proteins in the DNA repair and the maintenance of genomic stability. It has been hypothesized that genes coding for these proteins would be alternative candidates for ovarian cancer susceptibility. The Cancer Genome Atlas (TCGA) has showed that around half of high-grade serous ovarian cancers have aberrations in homologous recombination repair (See Fig. 7.1) [138, 139]. These patients with mutation of HRD-related gene are at increased risk for both ovarian and breast cancers, similar to BRCA1/2 mutation carriers. In addition, these tumors present a specific phenotype similar to BRCA-related ovarian cancers [7], including sensitivity to platinum agents and improved survival rates [71, 72]. The survival was similar for women with mutations in BRCA1 and other HRD-related genes (Fig. 2.2) [22].
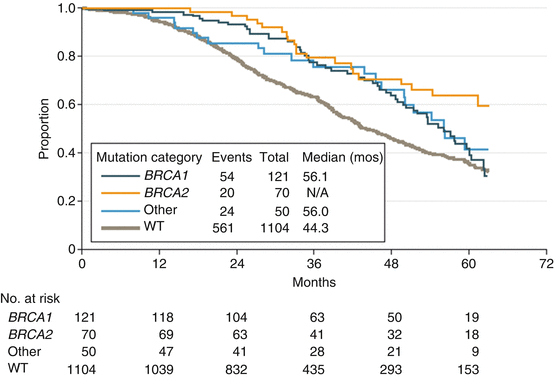
Fig. 2.2
Overall survival by mutation category in advanced ovarian cancers. The survival was similar for women with mutations in BRCA1 and other HRD-related genes in GOG 218 and GOG 262. GOG indicates Gynecologic Oncology Group; NA indicates not applicable; other indicates the genes BRIP1, PALB2, RAD51C, RAD51D, and BARD1; WT indicates wild type [22]
RAD51 genes are involved in homologous recombination, and this biallelic mutation can cause a Fanconi anemia-like phenotype [140]. RAD51C and RAD51D have been shown to be associated with increased risk for ovarian cancer [140]. In 1915 unselected ovarian cancer cases, 1.1% of patients had either a RAD51C or RAD51D mutation [22]. In cases from 1100 German families with gynecological malignancies, Meindl et al. identified six monoallelic pathogenic mutations in RAD51C that confer an increased risk for breast and ovarian cancer [141]. Loveday et al. reported that 8 inactivating RAD51D mutations were identified in unrelated individuals from 911 breast-ovarian cancer families, and the mutations confer a 6.3-fold increased risk of ovarian cancer but cause only a small increase in breast cancer risk (RR = 1.32) [142]. The analyses from the same trial including 1132 probands with a family history of ovarian cancer and 1156 controls also showed that RAD51C was associated with an increased risk for ovarian cancer (RR, 5.88; 95% CI, 2.91–11.88; P < 0.001) [143]. In a case-control analysis of 3429 ovarian cancer cases and 2772 controls, both RAD51C (OR, 5.2; 95% CI, 1.1–24; P = 0.035) and RAD51D (OR, 12.0; 95% CI, 1.5–90; P = 0.019) were associated with an increased risk for ovarian cancer [144]. The NCCN Guidelines recommend that RRSO in RAD51C and RAD51D mutation carriers is considered beginning at ages 45–50; however, further analyses are needed to confirm recommendation age of RRSO in these mutation carriers [13].
BRIP1, BRCA1-interacting protein C-terminal helicase 1, is a DNA helicase and defective in Fanconi anemia complementation group J. In 1915 unselected ovarian cancer cases, 1.4% of patients had a mutation in BRIP1 [22]. In analysis of Icelandic 656 ovarian cancer cases and 3913 controls, BRIP1 frameshift mutation confers an increase in ovarian cancer risk (OR, 8.13; 95% CI, 4.74–13.95; P < 0.001) [145]. In addition, an analysis of 3236 invasive ovarian cancer patients, 3431 controls, and 2000 unaffected high-risk women from a clinical screening trial of ovarian cancer (UKFOCSS) showed that BRIP1 is associated with a significant increased risk for ovarian cancer and relative risks associated with BRIP1 mutations were 11.22 for invasive ovarian cancer (95% CI, 3.22–34.10; P < 0.001) and 14.09 for high-grade serous disease (95% CI, 4.04–45.02; P < 0.001) [146]. The cumulative lifetime risk of developing ovarian cancer by age 80 in BRIP1 mutation carriers is estimated to be 5.8% (95% CI, 3.6–9.1) [146]. The NCCN Guidelines recommend that RRSO in BRIP1 mutation carriers be considered beginning at ages 45–50; however, their cumulative risk exceeds that of a woman with a first-degree relative with a non-BRCA-related ovarian cancer in around age 50–55 years. Further prospective trials are needed to confirm recommendation age of RRSO in these mutation carriers [13].
PALB2, partner and localizer of BRCA2, is a Fanconi anemia gene and an integral component of the BRCA complex required for homologous recombination repair [147]. PALB2 mutations have been detected in 1–4% of families negative for BRCA mutations [148]. Norquist et al. reported that 12 patients had germline mutations of PALB2 in analysis of 1915 ovarian cancer patients [22]. In sequence analysis of genomic DNA of 1144 familial breast cancer patients with wild-type sequences at BRCA1 and BRCA2, PALB2 heterozygotes were 1.3-fold more likely to have a relative with ovarian cancer (P = 0.18) [6]. Overall, significantly less ovarian cancer is seen in PALB2 families when compared with BRCA1 and BRCA2 families; therefore, it remains to be seen whether ovarian cancer risk is truly increased in individuals who are PALB2 mutation carriers or not [148].
2.4 PARP Inhibitors
Poly (ADP-ribose) polymerase (PARP) inhibitors cause cancer cell death in BRCA-mutated cancers by synthetic lethality. Olaparib was the first PARP inhibitor approved in the European Union and the USA for the treatment of advanced ovarian cancer patients with a germline BRCA mutation. The FDA approved olaparib for the patients who have received treatment with three or more lines of chemotherapy [149, 150]. Recent data suggest that olaparib is especially active in patients with platinum-sensitive recurrent ovarian cancer; on the other hand, a lower response rate is observed in patients showing resistance or refractory to platinum agent [151–156].
Related posts:
 Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
Ovarian Cancer Genome and Molecular Experimental Sciences
Strategies for the Management of Epithelial Ovarian Cancer
 Strategies for the Management of Non-epithelial Ovarian Tumors
Strategies for the Management of Non-epithelial Ovarian Tumors
 Morphological and Molecular Pathogenesis of Epithelial Ovarian Tumors
Morphological and Molecular Pathogenesis of Epithelial Ovarian Tumors
 Pathology of Epithelial Ovarian Tumors
Pathology of Epithelial Ovarian Tumors
 Pathology of Non-epithelial Ovarian Tumors
Pathology of Non-epithelial Ovarian Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


