Hereditary Hypertriglyceridemias
Lipoprotein lipase deficiency (familial hyperchylomicronemia)
Lipoprotein lipase (LPL) deficiency (OMIM No. 238600) is a rare autosomal recessive disorder, which was first reported as a separate entity in Europe by Bürger and Grütz in 1932 (hepatosplenomegalic lipidosis with xanthomatosis, the Bürger-Grütz disease), and by Holt and co-workers at Johns Hopkins University in 1939 (idiopathic familial lipemia) when the recessive mode of inheritance was recognized. The link with defective clearance of triglyceride-rich lipoproteins was made in the early 1960s by Havel and Gordon. It was also called familial fat-induced hyperlipemia because dietary fat markedly worsened the hypertriglyceridemia, and familial hyperchylomicronemia or type I hyperlipoproteinemia in the Fredrickson classification (referring to the selective accumulation of chylomicrons in fasting plasma). It was given a name related to its etiology when it was shown to be secondary to LPL gene (LPL) mutations. The word chylomicronemia is sometimes used instead of hyperchylomicronemia because in normal fasting plasma, chylomicrons are usually absent or barely measurable. The term ‘chylomicronemia syndrome’ has a more general meaning, referring to the presence in plasma of large lipoprotein particles that originate from dietary fat. This occurs in many forms of hereditary or acquired hypertriglyceridemia.
LPL is a key enzyme in lipid metabolism. It is expressed in various tissues, primarily adipocytes, skeletal muscle cells and the heart. Its primary function is to hydrolyse the triglyceride-rich lipoproteins (TRL), chylomicron and very-low-density lipoproteins (VLDL) in the vessel lumen to liberate monoglycerides and free fatty acids (FFA) for tissue utilization. In the process, smaller cholesteryl ester enriched remnant lipoproteins are formed for clearance by the liver and surface remnants carrying apolipoprotein AI (apoAI) are transferred to high-density lipoproteins (HDL). When this enzyme is deficient there is reduced formation of chylomicron remnants, fewer FFA reaching the liver to contribute to VLDL formation with fewer apoAI surface remnants for HDL, which results in high triglyceride, low low-density lipoprotein-cholesterol (LDL-C) and low HDL-C (2.1). The major co-factor and activator of LPL is apoCII present in HDL, VLDL and chylomicrons. FFA are the main energy source of skeletal muscle, and are stored in human adipose tissue if not used immediately for energy. The remnants of these TRL are eventually taken up by the liver through receptor-mediated endocytosis where LPL, apoE and proteoglycans contribute to ligand binding (2.2). The LDL receptor (LDLR) and the LDL receptorrelated protein (LRP) are the main receptors for remnant lipoprotein uptake in the liver. The VLDL receptor appears to be involved in other tissues (heart, skeletal muscle, fat, brain and macrophages) and the apoB-48 receptor located on macrophages may take up chylomicron remnants and favour foam cell formation. The existence of LPL was first recognized by Paul Hahn in 1943 when he noticed that an intravenous injection of heparin could clear postprandial lipemia in dogs. It was therefore given the name ‘heparinreleasable clearing factor’ because LPL, which is attached to the endothelium by membrane-bound heparan sulphate proteoglycan chains (2.2), is displaced by heparin that competes for binding to HSPG and released for massive action. Post-heparin lipolytic activity is used to demonstrate low or absent LPL activity in familial hyperchylomicronemia. The contribution of the two components, LPL and hepatic lipase (HL) can also be determined individually.
The human LPL gene is located on chromosome 8 (8p22), spans ∽30 kb, comprises 10 exons and encodes a 475 amino acid protein containing 5 domains including a 27-amino acid signal peptide. Nearly 100 different mutations have been found in the LPL gene, most located in exons 5 and 6, some of which affect the function of the enzyme. The most common mutations are the Asp9→Asn (D9N), Gly188→Glu (G188E), Asn291→Ser (D291S) and Ser447→Ter (S447X) substitutions (2.3). One mutation, Ser447→Ter (S447X), is associated with a gain of function of LPL activity, reduced triglycerides and increased HDL-C. LPL deficiency occurs with a frequency of about 1 in 1 000 000 worldwide. The frequency of the disease may be higher in areas where a founder effect exists. Homozygosity and compound heterozygosity for mutations in the LPL gene resulting in loss of enzyme function are the major causes of familial hyperchylomicronemia. Although LPL is bound to the capillary endothelium, LPL mRNA is not found in the endothelial cells but rather in the adjacent cells (i.e. hepatocyte, adipocyte, cardiomyocyte, or macrophage) from where it is secreted.
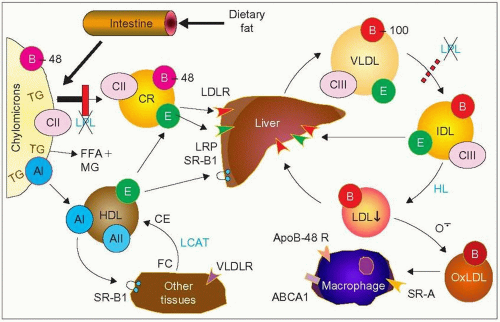 2.1 Metabolic defect in lipoprotein lipase (LPL) deficiency. Dietary fat is processed into micelles and absorbed in the intestine where it is packaged by the enterocytes into chylomicrons. These very large lipoprotein particles (>75 nm) are rich in triglycerides (TG) and maintained in ‘solution’ in plasma by their surface apolipoproteins, mainly apoB-48, apoAI and apoCs. ApoCII is an activator of LPL, surface apoAI is transferred to high-density lipoproteins (HDL) and apoB-48 serves to interact with receptors once these particles are broken down by LPL and acquire apoE from HDL to become chylomicron remnants (CR). This transformation is the result of the action of LPL bound to the endothelium that hydrolyses triglycerides into free fatty acids (FFA) – also referred to as non-esterified fatty acids (NEFA) – monoglycerides, and glycerol as the particles travel in the blood stream. The remnant particles are taken up by the liver through receptor-mediated endocytosis; essentially via the LDL receptor and the LDL receptor-related protein (LRP). LPL contributes to this receptor uptake. In LPL deficiency (shown by the red vertical bar) there is reduced formation of chylomicron remnants, less FFA reaching the liver to contribute to VLDL formation, and less apoAI surface remnants for HDL, which results in high triglycerides, low low-density lipoprotein-cholesterol (LDL-C) and low HDL-C. There is an apoB-48 receptor on macrophages that can take up chylomicron remnants and contribute to the formation of foam cells and therefore to atherogenesis. In LPL deficiency, fewer CM particles reach the macrophage apoB-48 receptor which may account for the rarity of atherosclerosis reported in this disease. LPL also contributes to the lipolysis of very-low-density lipoproteins (VLDL) and it is a paradox (still unexplained) that familial hyperchylomicronemia is usually not associated with an increase in VLDL. VLDL production can increase for other reasons, however; in this case the lipoprotein phenotype includes both an excess of chylomicrons and an excess VLDL (type V pattern) (see 2.7). The VLDL receptor (VLDLR) is not present in the liver and has been claimed to contribute to the uptake of lipoprotein remnants. See 1.2 for abbreviations and further information. |
LPL deficiency usually starts in childhood with symptoms of severe abdominal pain, acute pancreatitis, and hepatosplenomegaly, and failure to thrive. In adults, other typical manifestations include eruptive xanthomas (2.4), lipemia retinalis (2.5), memory loss, and sensory peripheral neuropathy. It is also characterized by severe fasting hypertriglyceridemia, usually greater than 11.3 mmol/l (1000 mg/dl); a large increase in chylomicrons, giving a milky appearance to plasma, which is topped by a creamy layer on standing; low HDL-C levels; and small dense LDL particles (2.6). These patients generally do not seem to be at increased cardiovascular risk.
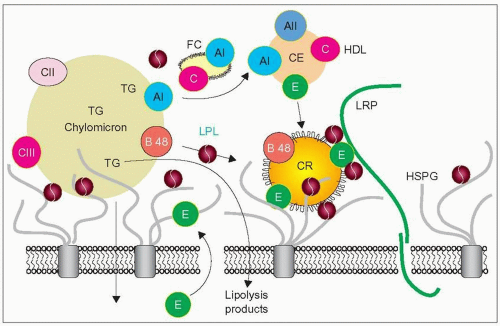 2.2 A model of chylomicron remnant uptake. The lipoprotein lipase (LPL) homodimers (dark red circles) can bind simultaneously to heparan sulphate proteoglycan (HSPG) (grey chains anchored to integral membrane proteins), to lipoproteins, and to other proteins such as apoE and receptors. This allows contact of several LPL molecules with the large chylomicrons for their effective hydrolysis. Lipolysis occurs through a series of attachment and detachment events. The products of lipolysis diffuse in the subendothelial space. ApoE (green circles) is secreted by the hepatocytes into the space of Dissé and reaches the endothelium surface to link with the remnants. LPL, because of its non-catalytic bridging function, facilitates apoE ligand binding to LDL receptor-related protein (LRP) and other receptors, thereby enhancing their clearance by the underlying hepatocytes. HSPG and apoE play a critical role in the sequestration and capture of the remnants. As the large particles shrink down into the remnant size, their surfaces become redundant (frills on CR) and surface components including free cholesterol (FC) and apoAI split off and are transferred to HDL. Chylomicron remnants (CR) gain apoE mostly from HDL, lose apoCII, and are taken up by LRP for endocytosis. It seems that hepatic lipase also secreted by the hepatocytes has a similar role to that of LPL. A direct uptake by HSPG has also been postulated. Composite diagram redrawn from Olivecrona T, Bengtsson-Olivecrona G (1993). Lipoprotein lipase and hepatic lipase. Curr Opin Lipidol, 4: 187-196. Ji ZS, Fazio S, Lee YL, Mahley RW (1994). Secretion-capture role for apolipoprotein E in remnant lipoprotein metabiolism involving cell surface heparan sulfate proteoglycans. J Biol Chem, 269: 2764-2772 and Mahley RW, Ji ZS (1999). Remnant lipoprotein metabolism: key pathways involving cell- surface heparan sulfate proteoglycans and apolipopprotein E. J Lipd Res, 40: 1-16. |
The frequency of heterozygosity for the four most common mutations of the LPL gene in population-based studies ranges from 0.04% to 22%. The reduction in postheparin plasma LPL activity also varies greatly depending on the mutation, from -53% for Gly188→Glu to +4% for Ser447→Ter substitutions. Consequently, the average change in plasma triglycerides of heterozygous carriers ranges from +78% to -8% and the change in HDL-C from -0.25 to +0.04 mmol/l. The odds ratio for ischemic heart disease in heterozygous carriers of the Gly188→Glu mutation is 4.9, and only 0.8 for those bearing the Ser447→Ter mutation. Most of the heterozygous subjects do not have the typical clinical manifestations of the homozygotes, unless a secondary cause of hypertriglyceridemia (such as obesity, alcohol consumption, diabetes mellitus, pregnancy) or other hyperlipidemia -inducing gene defects that contribute to increased TRL synthesis are present.
Diagnosis of LPL deficiency is based essentially on the typical clinical picture, in particular eruptive xanthomas in adults, and hepatosplenomegaly and abdominal pain in children, evidence of consanguinity in the family and demonstration of fasting chylomicronemia. The exquisite sensitivity of plasma triglycerides to dietary fat is another clue (2.7), but a fat load test is usually unnecessary and should be done with caution, if at all, as it has the potential to provoke an attack of pancreatitis. The diagnosis may be confirmed by measuring the post-heparin plasma LPL lipolytic activity, measuring post-heparin plasma LPL mass, or by more specifically identifying the causal mutation in the LPL gene, as may be done in several lipid clinics. Other primary and secondary causes of chylomicronemia should be excluded. The latter includes: acute alcohol misuse, primary acute pancreatitis with secondary hyperlipidemia, uncontrolled diabetes, hyperlipemia of pregnancy, plasma sample taken after a fatty meal, and mixed hypertriglyceridemia (type V lipoprotein phenotype) without LPL abnormality. Importantly, familial chylomicronemia may present on occasion as mixed hypertriglyceridemia, i.e. high exogenous and endogenous triglycerides (lipoprotein phenotype V), if one of these other conditions is present (2.7). This may happen in familial combined hyperlipidemia (see below). Interference with normal LPL activity is responsible for the other primary causes of hyperchylomicronemia, apoCII deficiency and familial hyperchylomicronemia, due to a circulating inhibitor of lipoprotein lipase.
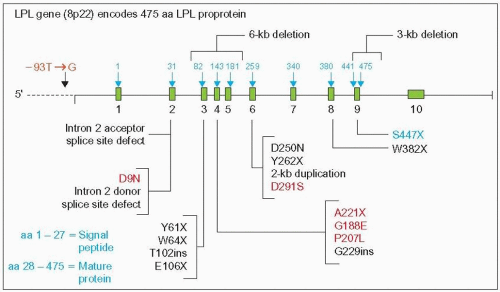 2.3 Major mutations of the lipoprotein lipase (LPL) gene. This diagram provides the gene location of a few of nearly 100 different reported mutations. The most common mutations of the LPL gene are depicted in red. Gilbert and co-workers ([2001]. Ann Génét, 4: 25-32) indexed all publications on LPL mutations and noted that one of them, G188E, was observed in 52 cases from 17 articles. These mutations are all associated with the chylomicronemia phenotype except S447X (blue), which is associated with an increased activity of LPL, reduced triglycerides, and increased high-density lipoprotein-cholesterol (HDL-C), and D291S, which has a modest effect on plasma lipoproteins. LPL mutations are distributed widely in the world. The promoter variant -93T→G represents 76% of LPL mutations in African blacks. The most common mutations among French-Canadians in the Province of Quebec where a founder effect exists are P207L, G188E, and D250N. Diagram partially updated from Santamarina-Fojo S (1992). Genetic dyslipoproteinemias: role of lipoprotein lipase and apolipoprotein C-II. Curr Opin Lipidol, 3: 186-195. |
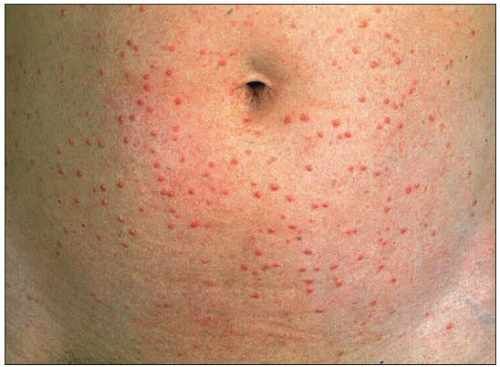 2.4 Eruptive xanthomas of the abdomen in lipoprotein lipase (LPL) deficiency. Eruptive xanthomas are usually yellowish to whitish, often with a reddish base. They are frequently found on the arms, back, thorax and buttocks. They appear in clusters, crops or as a few sparse lesions on hands or other parts of the body. They may develop within a few days and are often mistaken by the affected individual for a rash or an infection. They supervene in about 50% of subjects with LPL deficiency. |
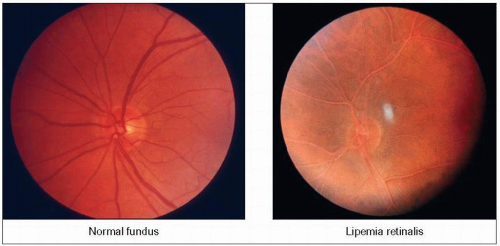 2.5 Lipemia retinalis in lipoprotein lipase (LPL) deficiency. In the normal fundus (left), it is easy to differentiate the pale arteries from the red veins. In contrast, in the lipemic fundus (right) there is virtually no difference in colour between the two. The entire retina and vessels have a pale salmon-pink discoloration. This is the fundus of a young woman with familial hyperchylomicronemia who is homozygous for the G188E mutation. Her lipoprotein electrophoreses are shown in 2.7. At the time this picture was taken she was hospitalized for acute pancreatitis and her plasma triglyceride level was 90.4 mmol/l (8000 mg/dl). The lipemic aspect of the retinal vessels proceeds from the periphery towards the centre gradually as triglycerides increase. The threshold for these changes is around 22.6 mmol/l (2000 mg/dl). |
ApoCII deficiency (OMIM No. 207750) is secondary to a mutation in APOC2 (2.8), the gene on chromosome19q13.2 that encodes apoCII, the activator of LPL. Breckenridge reported the first cases of apoCII deficiency in Canada in 1978, finding that a transfusion for anemia providing normal apoCII improved the associated severe hypertriglyceridemia. ApoCII deficiency essentially mimics the clinical and biochemical features of familial LPL deficiency except that affected subjects tend to be detected at a later age than the former (13-39 years of age, but as early as 6 years and as late as 60 years) and the phenotype is generally milder, probably depending on the level of residual apoCII in plasma. One case in infancy presented as a lipid encephalopathy. In patients with the disease, apoCII is reduced or absent on isoelectric focusing (IEF) gel electrophoresis of delipidated VLDL (2.9). The LPL defect is corrected in vitro by addition of plasma from the patient compared with that of a normal subject to activate lipase in the skim-milk lipase assay. Specialized laboratories can determine the specific mutation if necessary.
Familial hyperchylomicronemia due to a circulating inhibitor of lipoprotein lipase (OMIM No. 118830), has been reported in 1983 by Brunzell and co-workers, found in a mother and son. The mother had a clinical phenotype similar to that of LPL deficiency, with eruptive xanthomas, massive hypertriglyceridemia (20.4 mmol/l or 1813 mg/dl of chylomicron triglycerides), no diabetes and a history of unexplained abdominal pain in childhood. Her apoCII was normal. Unlike LPL deficiency, however, LPL was present in adipose tissue at 30-fold and two-fold greater levels than in normal subjects in mother and son, respectively. Their plasma contained an unidentified inhibitor of LPL capable of inhibiting post-heparin plasma lipolytic activity. Present in the non-lipoprotein fraction, the inhibitor was heat-stable, non-dialysable, and sensitive to repeated freezing and thawing. Inheritance did not appear to be recessive as the son and a grandson had marked hypertriglyceridemia. This anomaly remains an enigma.
Treatment of familial hyperchylomicronemia relies on drastic reduction of dietary lipids to less than 10-15% of total calories. This will reduce the triglyceridemia in a few days or weeks (2.7). Alcohol intake is contraindicated. Medium-chain triglycerides (MCT) are used to provide a source of fat that does not contribute to chylomicron formation. The latter are made in the intestinal wall from absorbed long chain fatty acids (≥16 carbon chains) and reach the circulation via lymphatics; MCT (12-14 carbon chains), in contrast, gain access to the liver via the portal vein and are metabolized differently. Lipid-lowering medications are ineffective in homozygous subjects. In the impending pancreatitis state, stopping food intake and maintaining hydration with water only for 1-2 days will alleviate abdominal pain and may avert an acute pancreatitis. Chylomicronemia syndromes are further discussed below (see Familial mixed hypertriglyceridemia).
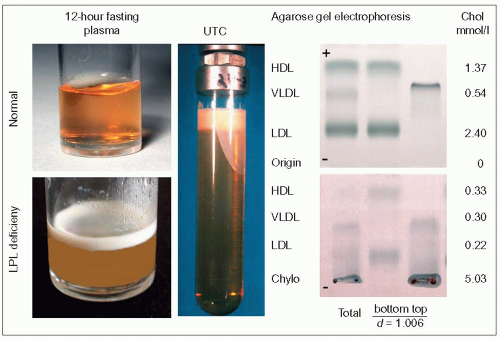 2.6 Demonstration of hyperchylomicronemia in lipoprotein lipase (LPL) deficiency. The left panel compares normal plasma and plasma from an LPL-deficient patient which were left to stand overnight in a refrigerator. The blood samples were obtained after a 12-hour fast. The floating creamy supernatant is typical of hyperchylomicronemia. The plasma before standing is turbid and whitish. The ‘refrigerator test’ is the simplest way to demonstrate the presence of chylomicrons for the practitioner. Ultracentrifugation at the aqueous density of plasma (d = 1.006), 50 000 rpm in a 50.4 Ti Beckman rotor for 8 hours developing 312 000 × g, separates chylomicrons and very-low-density lipoproteins (VLDL) at the top of the tube from the other plasma lipoproteins which sink to the bottom (centre panel). The creamy layer is chylomicrons; the white part adhering to the wall downward represents smaller particle size triglyceride-rich lipoproteins (TRL), mostly VLDL. This is the original preparative ultracentrifugation procedure developed by Havel et al. ([1955]. J Clin Invest, 35: 1345.) which separates lipoproteins according to their relative buoyancy. Total plasma and separated lipoproteins (top and bottom of the tube) can then be further separated according to charge by agarose gel electrophoresis as seen in the right panel. The migration is from the origin (-) upward towards the anode (+). The upper electrophoresis is that of a normal plasma, the lower one, that of a patient with LPL deficiency. In the latter, chylomicrons stay at the origin, and the low-density lipoproteins (LDL) (β-lipoproteins), VLDL (preβ-lipoproteins) and high-density lipoproteins (HDL) (α-lipoproteins) are very discrete bands (cholesterol values for these different fractions are given on the right). There are no chylomicrons in normal fasting plasma. The same amount of plasma was deposited at the origin in both cases. Chylomicrons are buoyant and separate readily without having to resort to prolonged ultracentrifugation. Centrifugation at 58 450 × g will allow their separation from plasma in 30 minutes.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|