Fig. 20.1
Distribution of colorectal cancer cases (Courtesy of Quyen D. Chu, MD, MBA, FACS)
Lynch Syndrome
The most common form of heritable CRC is Lynch syndrome, which accounts for 1–3 % of all cases of CRC [4, 5]. It is inherited in an autosomal-dominant fashion. Lifetime risk of CRC in Lynch syndrome is approximately 10–69 % depending on gender and mismatch repair gene mutations [6–8]. Lynch syndrome is used in preference to hereditary nonpolyposis colorectal cancer (HNPCC) given that polyps can occur, and it involves a group of extracolonic cancer types.
Genetics
Lynch syndrome is characterized by a mutation in one of the DNA mismatch repair (MMR) genes – MLH1, MSH2, MSH6, and PMS2 (Fig. 20.2). These genetic mutations lead to errors in the number of repetitive sequences replicated, causing microsatellite instability (MSI). The errors that occur during DNA replication are not efficiently repaired, causing mutant changes and subsequent unrestrained growth that leads to adenoma and then to carcinoma. MSI occur in approximately 90–95 % of cancers in Lynch syndrome due to uncorrected errors in DNA replication [3].
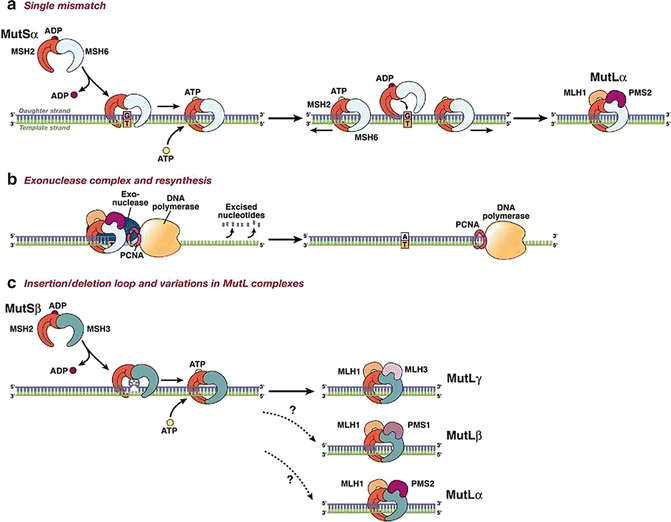
Fig. 20.2
The DNA MMR system functions through a series of steps. (a) MSH2–MSH6 (MutSα) recognizes single base-pair mismatches, in which the DNA polymerase has matched the wrong base (G) with the T on the template (shown on left), and creates a sliding clamp around the DNA. This step that requires the exchange of adenosine triphosphate (ATP) for adenosine diphosphate (ADP) (by MSH2, but not MSH6 or MSH3). The complex diffuses away from the mismatch site, which is then bound by the MLH1–PMS2 (MutLα) complex (right). This “matchmaker” complex moves along the new DNA chain until it encounters the DNA polymerase complex. (b) The DNA MMR protein sliding clamp interacts with exonuclease-1, proliferating cell nuclear antigen (PCNA), and DNA polymerase. This complex excises the daughter strand back to the site of the mismatch (shown on left). Eventually, the complex falls off the DNA and resynthesis occurs, correcting the error. (c) Variations on the DNA MMR theme. Whereas MSH2–MSH6 recognizes single-pair mismatches and small IDLs, MSH2–MSH3 (MutSα) complements this by also recognizing larger IDLs (shown on left). The right side shows the possible interactions with different MutL dimers, as MLH1 can dimerize with PMS2, PMS1, or MLH3. The preferred interaction with MSH2–MSH3 is MLH1–MLH3 (MutLα), but the precise roles of the other MutL heterodimers in this reaction are not entirely understood (Reprinted from Ref. [128]. With permission from W.B. Saunders Co.)
In a report published by the International Collaborative Group on HNPCC, 63 % of total mutations reported in Lynch patients were MLH1 mutations, 25 % were MSH2 mutations, 6 % were MSH6 mutations, and 0.4 % were PMS2 mutations [3]. No clear genotype–phenotype relationship has been established except in Lynch patients that present with endometrial cancer, which is most commonly associated with MSH6 mutations [3]. However, patients with mutations in the other MMR genes can develop endometrial cancer.
Sometimes, patients will display mutations in mismatch repair proteins or high microsatellite instability (MSI-H) without evidence of germline mutations. This can be due to current technology that cannot identify the mutations. Also, deletions in epithelial cell adhesion molecule gene (EpCAM), also known as TACSTD1, located just upstream of MSH2 can account for Lynch syndrome [9]. This mutation leads to hypermethylation of MSH2 that can ultimately lead to an EpCAM–MSH2 fusion protein that can cause aberrant protein transcription. This is responsible for the Lynch phenotype in 6–19 % of families without MMR gene mutation [10].
Clinical Evaluation
A detailed family history of at least three generations should be obtained in all patients being evaluated for Lynch syndrome. Clinical criteria such as the Amsterdam I and II were developed to identify high-risk families to aid in the discovery of the MMR genes. These criteria are useful and are still being used to identify Lynch syndrome kindreds. Lynch syndrome is suspected in those that fit the Amsterdam II criteria [11]. This criteria requires that at least three relatives, of which one must be a first-degree relative of the other two, have a diagnosis of some cancers associated with Lynch syndrome (CRC, endometrial, ureter/renal pelvis, small bowel), that at least two successive generations be affected, that at least one relative had a diagnosis of cancer associated with Lynch syndrome before the age of 50, and where FAP has been excluded. The Bethesda guidelines [12] linked the diagnostic criteria of Lynch syndrome to the presence of microsatellite instability (Table 20.1). However, with the advent of molecular technologies such as polymerase chain reaction (PCR) and immunohistochemistry for MMR gene protein expression in colorectal tumors (and in some cases extracolonic tumors), neoplasms can be screened for mismatch repair deficiency (and proficiency) thus identifying individuals that would be missed if only clinical criteria were used for guidance in identifying Lynch syndrome patients.
Table 20.1
Amsterdam II criteria/Bethesda guidelines
Greater than 3 relatives with Lynch/HNPCC-associated cancer and |
1. One should be a first-degree relative of the other two |
2. At least two successive generations should be affected |
3. At least one diagnosed before the age of 50 |
4. FAP should be ruled out |
5. Tumors verified by pathological examination |
Tumors should be tested for microsatellite instability in following situations: |
1. Colorectal cancer under age of 50 |
2. Presence of synchronous, metachronous colorectal, or other HNPCC-related cancers |
3. Colorectal cancer with high microsatellite instability histology diagnosed in patients below age of 60 |
4. Colorectal cancer diagnosed in patient with one or more first-degree relatives with an HNPCC-related tumor confirmed under age of 50 |
5. Colorectal cancer diagnosed in patient with two or more first-degree relatives with an HNPCC-related tumor confirmed at any age |
Extracolonic cancers include endometrial, gastric, urinary tract, pancreas, biliary tract, brain, sebaceous gland adenomas, keratoacanthomas in Muir–Torre syndrome, carcinoma of the small bowel, and ovarian neoplasias [13, 14].
Muir–Torre variant of Lynch syndrome is associated with dermatologic manifestations such as sebaceous adenomas and carcinomas, keratoacanthomas, and basal carcinomas with sebaceous differentiation in addition to the other Lynch-associated tumors [15].
Surveillance
Those patients that meet the Bethesda guidelines should be offered screening by MSI testing or by immunohistochemistry to look for loss of MMR protein expression. However, the Bethesda guidelines were sensitive but not specific enough. There has been a move toward universal testing of colorectal tumors because it will help identify patients with Lynch syndrome as well as the status of the mismatch repair (either proficient or deficient) [16]. A more selective approach for testing colorectal cancers has been recommended by Moreira et al. and the Epicolon consortium [17]. These authors recommend testing all CRC diagnosed at age 70 or less and in older patients who fulfill the revised Bethesda guidelines. Using this approach, 4.9 % of Lynch syndrome cases were missed, but 34.8 % fewer cases required tumor MMR testing and 28.6 % fewer patients underwent germline mutation testing compared to a universal approach to all colorectal tumors [17]. It must be emphasized that tumors that show loss of protein expression of MLH 1 should undergo BRAF mutation and/or methylation testing of the promoter of MLH1. BRAF mutations are demonstrated in high levels in sporadic MSI CRC and rarely in Lynch syndrome CRC [18] (Fig. 20.3). Those patients whose tumors display loss of MSH2 protein are considered to have Lynch syndrome either secondary to the mutation in MSH2 or less commonly a mutation in EpCAM causing epigenetic silencing of MSH2. Less commonly isolated loss of either PMS2 and MSH6 will be noted directing the clinician to test for germline mutations in these genes. Most commonly loss of MSH6 is accompanied by loss of MSH2 and loss of PMS2 by loss of MLH1.
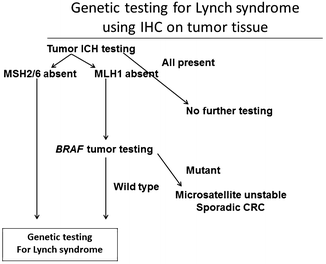
Fig. 20.3
Algorithm for genetic testing for Lynch syndrome. CRCs are tested via immunohistochemistry first for presence or absence of DNA mismatch repair proteins. If all proteins are present, then Lynch syndrome is ruled out. If MLH1 is absent, then the tumor is analyzed for BRAF mutations. If BRAF protein is present in its original state or if MSH 2 or 6 is absent, then the patient is tested genetically for Lynch syndrome. If BRAF protein is present as a mutant state, then CRC is likely a sporadic tumor due to microsatellite instability (Reprinted from Ref. [129]. With permission from Nature Publishing Group)
Annual full colonoscopy starting at age 20–25 is recommended for those with diagnosis of Lynch syndrome3. Strong clinical evidence suggests more rapid transition from adenoma to carcinoma in those with Lynch syndrome, thus a more frequent endoscopic surveillance than for the general population is warranted. Colonoscopic surveillance in Lynch syndrome has been shown to decrease CRC incidence and decrease mortality from CRC [19].
Some literature advocates for annual transvaginal ultrasonography, measurement of CA 125 levels and annual endometrial aspiration for affected females starting at age 25–35 [20]. In those patients with Lynch syndrome and family history of gastric cancer, annual EGD is recommended. Ultrasonography and urine cytology can be considered annually or every other year to screen for urinary tract malignancy. Data for these screening tools to decrease mortality is limited at best.
Surgical Treatment
Due to a high lifetime risk of CRC, prophylactic surgical options should be presented to patients diagnosed with Lynch syndrome. However, if the colon is normal in appearance on colonoscopic exam, surgery is not recommended unless there are extenuating circumstances. The options of treatment in Lynch syndrome include total abdominal colectomy with ileorectal anastomosis (IRA) with yearly flexible sigmoidoscopy in those with normal rectal and anal sphincter function or segmental colectomy with yearly colonoscopy. To date, there have been no prospective or retrospective studies demonstrating a survival improvement in patients undergoing a total abdominal colectomy versus a segmental colectomy. What has been demonstrated is a decrease in metachronous colorectal cancer and abdominal procedures related to CRC in patients undergoing more extensive procedures [21, 22]. In the study by Parry et al., the risk of metachronous CTC after a segmental colectomy was 16 %, 41 %, and 62 % at 10, 20, and 30 years after segmental resection in MMR mutation carriers, respectively. Careful surveillance should also be advocated for those that opt for IRA, since the risk of metachronous rectal cancer after total colectomy was reported to be approximately 12 % at 10–12 years [23]. Because of the risk of metachronous CRC, most recommend a total abdominal colectomy at the time of diagnosis of colon cancer in Lynch syndrome. If the index cancer is in the rectum, the alternatives include segmental resection versus restorative proctocolectomy with ileal pouch-anal anastomosis (IPAA) if the sphincters are not involved. Similar to the colon, there is an increased incidence of metachronous colon cancer in patients undergoing segmental rectal resection. These have been reported to be 19%, 47% and 69% at 10, 20, and 30 years post-segmental rectal resection in mutation carriers [24].
Extracolonic Manifestations
Endometrial cancer risk has been reported to be from 15 % to 71 % in MMR gene carriers [6–9, 25]. Endometrial cancer can be the index cancer in a Lynch syndrome patient. Similar to CRC, the age of presentation is younger than the general populations. There is retrospective data reported where females who underwent prophylactic hysterectomy and salpingo-oophorectomy did not develop endometrial or ovarian cancer. In those that did not have the prophylactic procedure, 61 out of 315 females developed cancer [26]. In patients who have completed their families or are postmenopausal, discussion about prophylactic hysterectomy and salpingo-oophorectomy should be entertained, especially at the time of colectomy for CRC.
Other extracolonic manifestations associated with Lynch syndrome include gastric, urinary tract, pancreatic, biliary, brain cancers, sebaceous glands, and keratoacanthomas. There has been a suggestion that both prostate and breast cancer are part of the tumor spectrum of Lynch syndrome, but because these tumors are so common in the general population, there is still controversy about their link to Lynch syndrome [24].
Familial Adenomatous Polyposis
Familial adenomatous polyposis (FAP) is the second most common inherited colon cancer, affecting approximately 1 in 10,000 individuals and accounting for approximately 1 % of all colon cancers [27]. It is inherited in an autosomal-dominant fashion with nearly 100 % penetrance. Those affected are at nearly 100 % risk of CRC by the age of 60 [3].
Genetics
Adenomatous polyposis coli (APC) gene is a tumor suppressor gene that spans 108 kb of DNA on chromosome 5q21. The gene encodes a protein that negatively regulates the β-catenin oncoprotein. In the absence of the APC gene, the β-catenin protein interacts with various transcription factors as it accumulates in the nucleus to upregulate genes that propagate the cell cycle progression [28]. Germline mutations include deletions, insertions, nonsense, and missense mutations. Overall, it is predicted that a mutant truncated APC protein is produced as a consequence of these mutations in as much as 92 % of the cases [29] (Fig. 20.4).
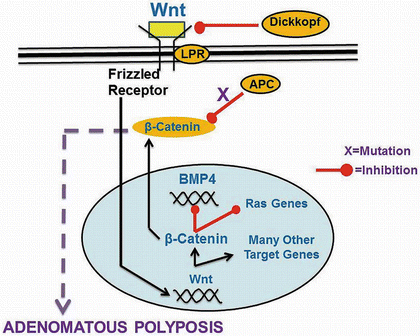
Fig. 20.4
Wnt/β-catenin pathway and adenomatous polyposis. APC (adenomatous polyposis coli) normally targets β-catenin for degradation. In familial adenomatous polyposis and in sporadic adenomatous polyps, mutations in APC are associated with an increase in β-catenin (Reprinted from Ref. [130]. With permission from Springer Verlag)
Majority of the germline mutations are clustered in the 5′ portion of exon 15. Miyoshi and colleagues reported that 40 % of all mutations occurred in five specific codons (302, 625, 1061, 1309, and 1546), and 65 % of somatic mutations and 23 % of germline mutations in FAP patients occurred between codons 1286 and 1513 in exon 15 [30]. Genotype–phenotype correlation studies have demonstrated that severe polyposis in FAP patients usually have mutations between codons 1250 and 1464. Correlation between severe FAP and earlier age of onset (defined as symptoms in teen years, cancer before 30), higher number of polyps, and higher mean of diagnosis and death were seen in those with codon 1309 mutations and those more downstream [31–33]. However, subsequent studies have shown more heterogeneity and variability between and within family members with FAP and codon 1309 mutations, showing that a specific APC mutation was not the only determinant of phenotype [34].
Clinical Evaluation
Classically, FAP is diagnosed in those with greater than 100 adenomatous colorectal polyps. Polyp development is usually evident around puberty. In a review of a national polyposis registry, median age for development of colorectal adenomatous polyps was 16 years (range 5–38 years), first symptoms from lower GI tract developed by median age of 29 years (range 2–73 years), and development of colorectal carcinoma occurred at a median age of 36 years (range 17–67 years) [3]. Earliest symptoms and signs of lower GI involvement in patients with FAP are blood per rectum, vague abdominal pain, tenesmus, diarrhea and/or constipation, or obstipation.
Polyps develop by age 20 in 75 % of cases and are usually less than 1 cm in size [3]. They may be pedunculated or sessile and may have tubular, villous, or tubulovillous histology. In severe polyposis, thousands may carpet the colorectal epithelium. The risk of invasive cancer is proportional to the severity of polyposis. CRC in the setting of FAP tends to be more commonly located on the left side, unlike CRC in the setting of Lynch syndrome [4].
A number of extracolonic manifestations have been reported in patients with FAP. Those include desmoid tumors, periampullary neoplasms, osteomas, odontomas, supernumerary teeth, fused teeth roots, sebaceous and epidermoid cysts, hepatoblastomas, thyroid tumors, and congenital hypertrophy of the retinal pigmented epithelium (CHRPE).
Surveillance
Patients in whom the diagnosis of FAP is suspected should undergo a complete history, paying particular attention to family history and physical examination with emphasis in areas affected by extracolonic manifestations, including neurologic, ophthalmic, dental, dermatologic, thyroid, abdominal, and digital rectal examinations.
If a mutation is known in the family, then the at-risk individual(s) should be tested for that mutation. In general, genetic testing is not recommended before age 10–12 years. Patients with normal gene study can be dismissed from further screening with a nearly 100 % certainty that any known mutation is absent. These patients should still be counseled to undergo CRC screening starting at age of 50, which is the recommendation for the general population. Surveillance for at-risk family members should begin at 10–12 years of age with a flexible sigmoidoscopy, repeated at 1–2 year intervals. Those that present with adenomatous polyps should undergo a full colonoscopy to determine the extent of polyposis. It is understood that if there are any symptoms prior to the recommended age of surveillance, at-risk individuals should be immediately evaluated at the onset of symptoms.
About 25 % of patients with FAP have no family history of polyposis; therefore, the mutation is de novo [35]. Grover et al. reported the prevalence of germline mutations in the APC gene according to the number of adenomas [36]. The prevalence of APC germline mutation in patients with 10–19 adenomas was 5 %, whereas if there are greater than 1,000 adenomas, it was 80 %.
Treatment
Surgical
Due to the nearly 100 % risk of colorectal cancer, prophylactic surgery has become the standard of care in patients with FAP. The timing of surgical treatment partially depends on the age of the patient, extent of polyposis, symptoms, family history of desmoids, genetic test results (if available), experience of the surgeon, and patient’s input. The risk of CRC in FAP patients less than 20 years of age is less than 1 % [37]. If a prophylactic colectomy is to be performed due to severe polyposis, then ideally it should be performed between high school and college. Patients with severe polyposis, severe dysplasia, adenomas greater than 5 mm, and those with severe symptoms that impair quality of life should undergo surgery as soon as possible [38]. Prophylactic colectomy may also be delayed in those with a family history of aggressive desmoids tumors because the risk of desmoids-related complications may outweigh the risk of developing CRC. In females, Ileal pouch anal anastomosis (IPAA) has been associated with decreased fecundity. Therefore, it is also reasonable to delay surgery if deemed safe [39].
The three basic surgical options are: (1) total proctocolectomy (TPC) with permanent ileostomy, (2) total abdominal colectomy with ileorectal anastomosis (IRA), or (3) restorative proctocolectomy (with or without mucosectomy) with ileal pouch-anal anastomosis (IPAA).
TPC with permanent ileostomy is rarely chosen as the first-line option for prophylactic surgery. More commonly, it is considered when sphincter sparing surgery is not feasible due to rectal cancer presenting in the lower third of the rectum, if the patient has poor sphincter function or in the extremely rare situation of a patient presenting with desmoid disease that shortens the small bowel mesentery and not enough length can be technically achieved for an IRA or IPAA. Additionally, the patient’s lifestyle has to be taken into consideration.
The choice between IRA and IPAA is more challenging, and considerations for the risk of rectal cancer development and/or differences in functional outcome and quality of life must be taken into account. One of the advantages of an IRA is that it is a one-stage procedure, whereas IPAA, due to the creation of a diverting loop ileostomy to protect the distal anastomosis, is a two-stage procedure (although in expert hands, it can be a one-stage procedure). Besides the increased risk in morbidity that comes from an ileostomy takedown, IPAA has been cited by several studies to have increased rate of complications. Nyam et al. reported a complication rate of 24 % in 187 patients that underwent IPAA for FAP [39]. The most common complication reported was intestinal obstruction in 13 % of the patients. Other complications included wound infection, pelvic infection, urinary tract infection or retention, and sexual dysfunction. Later study by Kartheuser and colleagues reported similar findings, with a complication rate of 27 %. Approximately 15 % experienced small bowel obstruction, and approximately 14 % experienced other complications including pelvic sepsis, fistula formation, necrotizing enterocolitis, and anastomotic stricture (4 %). Impotence and retrograde ejaculation reported after IPAA has only been 1–3 %. More commonly seen is dyspareunia in females and stool leakage during intercourse [40].
A disadvantage with IRA is its association with rectal cancer risk. According to various studies, the risk of developing rectal cancer following IRA in patients with FAP is between 4 and 14 % after 10 years and 9 to 32 % after 20 years [41–45]. It is important to note that in some of these studies, figures were derived when only IRA was available even in the setting of more extensive rectal disease. Also clear documentation of the length of the rectal stump in some series was not provided. Therefore, rectal cancer occurrence after IRA may be overestimated. Iwama et al. noted that 3 % of patients with rectal stump shorter than or equal to 7 cm developed rectal cancer after IRA as opposed to 17 % in those with rectal stump longer than 7 cm [44].
The risk of developing rectal cancer after primary prophylactic surgery may be estimated on the basis of a specific location on the APC mutation. Those that had mutations downstream of codon 1250 had threefold higher incidence of rectal cancer than those with a mutation upstream of 1250 [46]. In another study, those that had an APC mutation between codons 1250 and 1464 were 6.2 times more likely to develop rectal cancer than those with mutations upstream of 1250 or downstream of 1464 [47]. Nevertheless, having APC mutations at other sites will not preclude potential rectal cancer after an IRA.
Furthermore, in a genotype–phenotype study, the cumulative risk of needing a secondary proctectomy within 20 years after IRA and the cumulative risk of developing rectal cancer were compared based on the location of the APC mutation. In the attenuated phenotype (discussed in separate section below) group, which correlated with codons 1–157, 312–412, and 1596–2843, the risks were 10 % and 3.7 %, respectively. In the intermediate phenotype group, correlating with codons 158–311, 413–1249, and 1465–1595, the risks were 39 % and 9.3 %, respectively. In the severe phenotype group, correlating with codons 1250–1464, the risks were 61 % and 8.3 %, respectively [48]. Church et al. correlated the number of rectal polyps at the time of prophylactic IRA with subsequent need for secondary proctectomy [49]. None of the patients with less than five rectal adenomas and less than 1,000 adenomas in the colon had to undergo proctectomy. Patients who had 5–20 rectal adenomas had a 13 % chance of subsequent proctectomy. Those with greater than 20 adenomas had a 54 % chance of subsequent proctectomy.
Patients that develop rectal cancer after IRA may undergo completion proctectomy. The rate of this procedure ranges from 36.6 % to 74 %. The 5-year survival rate following metachronous rectal cancer in FAP patients who had undergone IRA originally ranges from 60 % to 78 % [3]. Other factors that also influenced this rate were stage of the tumor, comorbidities, and performance status of the patient.
The risk of developing polyps and subsequent cancer is not limited to IRA. One report found the risk of developing polyps in the ileal pouch in patients who had undergone IPAA to be 7 % at 5 years, 35 % at 10 years, and 75 % at 15 years [50]. Another study reported a higher incidence of neoplasia occurring at the site of anastomosis in FAP patients who had undergone IPAA after staple use (31 %) versus those that received a hand-sewn anastomosis with anal mucosectomy (10 %).
In terms of bowel function and quality of life, stool frequency ranged from 4.5 to 5 after IPAA compared to 3–4 after IRA. Normal continence was reported in 60–87 % in patients after IPAA compared to 72–83 % after IRA [51, 52]. Night defecation was significantly less in the IRA group, although fecal urgency was reduced in the IPAA patients. Reoperation rate is higher in the IPAA group. No significant difference was shown in terms of sexual dysfunction, dietary restriction, or postoperative complications [53].
Regardless of the procedure, endoscopic surveillance is recommended at intervals of 6 months to 1 year, either to examine the rectal stump (in the case of IRA) or the ileal pouch (in the case of IPAA). After IRA, small adenomas less than 5 mm can be safely observed with biopsies taken. If adenomas increase in number, endoscopic surveillance should occur more frequently. Any polyps larger than 5 mm should be removed and examined by histology. Any development of dysplasia or a villous adenoma larger than 1 cm may be an indication for proctectomy if it cannot be addressed endoscopically. Chemoprevention is discussed in a separate section below.
Extracolonic Manifestations
Desmoid tumors are histologically collagen abundant, spindle cell populated benign tumors arising from fibroaponeurotic tissues. They are usually referred to as benign without metastatic potential but can be locally invasive with ill-defined margins3. The prevalence of desmoid tumors in FAP has been estimated to be as high as 38 % [54]. Desmoid tumors occur in approximately 10 % of FAP patients. One study estimated the cumulative risk of 21 % for patients with FAP to develop a desmoid tumor by age 60 [55]. Desmoid tumors are associated with high morbidity and can be the cause of death in 10–23 % of FAP patients [54]. A high proportion of desmoid tumors develop after colonic resection in FAP patients [46]. Patients that have had total colectomy with IRA more frequently developed intra-abdominal desmoids compared to those that of IPAA [56], and due to mesenteric shortening, IPAA may be technically impossible for patients needing a completion proctectomy after an initial IRA [37]. Females of reproductive age seem to be more prone to developing intra-abdominal desmoids [29], possibly due to the expression of estrogen receptors that have been shown in some desmoid tumors [57].
Pharmacological intervention has been used to treat desmoid diseases. NSAID therapy such as sulindac and indomethacin has shown to show partial to complete regression in small, non-randomized studies [58]. However, it is associated with significant side effects and delayed response. Hormonal agents such as tamoxifen have also shown variable partial or complete regression of desmoid tumors [59]. Either NSAIDs or hormonal agents are viable first-line options to treat clinically inert desmoid tumors. For fast-growing tumors or those unresponsive to NSAID or hormonal agents, cytotoxic agents such as doxorubicin and dacarbazine can achieve some degree of response [58–60]. There is no definite effective treatment for desmoid tumors. Church et al. proposed a classification system for desmoids in FAP patients that can serve as a guide for management of these difficult problems [61] (Table 20.2).
Table 20.2
Desmoid tumor staging system
Stages | |
|---|---|
I | Asymptomatic, less than 10 cm maximum diameter and not growing |
II | Mildly symptomatic, less than 10 cm maximum diameter and not growing |
III | Moderately symptomatic or bowel/ureteric obstruction or 10–20 cm or slowly growing |
IV | Severely symptomatic or greater than 20 cm or rapidly growing |
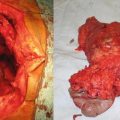
Surgical resection is limited to those that are symptomatic (e.g., from intestinal obstruction) (Fig. 20.5). Unfortunately, resection is associated with a high rate of and more aggressive recurrence.
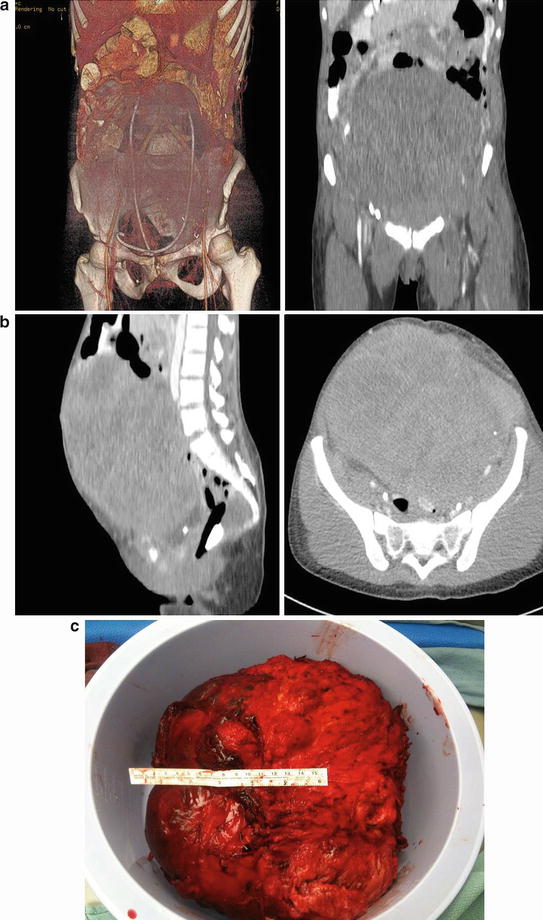
Fig. 20.5
A 24-year old woman with familial adenomatous polyposis presented with small bowel obstruction because of a large abdominal desmoids. She underwent a palliative debulking of her desmoids tumor (Courtesy of Quyen D. Chu, MD, MBA, FACS)
Duodenal cancer has become the leading cause of death in patients with FAP who have already undergone prophylactic colectomy [60]. Nearly 90 % of patients with FAP will develop duodenal polyps, and 4.5 % will develop duodenal adenocarcinoma in their lifetime [62].
Surveillance by EGD with biopsy of suspicious polyps should begin at age 20 or at the time of prophylactic colectomy, whichever is earlier [57]. Staging for duodenal polyposis can be staged using the Spigelman classification [63] (Table 20.3).
Table 20.3
Spigelman classification
Points | |||
|---|---|---|---|
1 | 2 | 3 | |
Polyp number | 1–4 | 5–20 | Greater than 20 |
Polyp size (mm) | 1–4 | 5–10 | Greater than 10 |
Histology | Tubular | Tubulovillous | Villous |
Dysplasia | Mild | Moderate | Severe |
Having no polyps is designated as stage 0. One to four points is classified as stage 1. For these stages, the surveillance should be every 5 years. Five to six points is stage 2, with surveillance recommended every 3 years. Seven to eight points is stage 3 with surveillance recommended every 1–2 years. Nine to twelve points is stage 4, which warrants surgical intervention.
Surgical options include endoscopic ablation and transduodenal excision. Duodenal surgery, specifically pancreas-preserving duodenectomy or pancreaticoduodenectomy, is currently indicated for patients with severe duodenal polyposis (Spigelman IV) or duodenal carcinoma.
Several small-powered studies have investigated the role of sulindac in stabilizing or regressing duodenal polyposis [64, 65]. So far, no significant benefits have been seen. A randomized placebo study using celecoxib showed that there was no significant difference among the groups in number of polyps, although there was significant qualitative improvement in polyposis among those on high-dose celecoxib when the patients’ endoscopies were reviewed independently by other physicians [66]. Overall, chemoprevention studies of duodenal polyposis with NSAIDs have been disappointing. One plausible explanation is that because the duodenum expresses higher levels of COX-2 than colon in FAP patients [67], higher dosage of NSAIDS may be needed to suppress polyp burden in the duodenum. However, higher dosages of NSAIDs, especially COX-2 inhibitors, may be limited by the potentially serious cardiovascular side effects.
Other extracolonic manifestations include gastric cancer, osteomas, odontomas, sebaceous and epidermoid cysts, and CHRPE (congenital hypertrophy of the retinal pigment epithelium).
Attenuated Familial Adenomatous Polyposis
A milder form of FAP known as attenuated familial adenomatous polyposis (aFAP) has been defined as less than 100 adenomatous polyps in the colon. Similar to FAP, aFAP is passed onto progeny by autosomal-dominant pattern and is associated with APC gene mutations and upper GI lesions [3]. Historically, aFAP has been reported to be predominantly right sided with rectal sparing, unlike FAP. However, recent studies indicate that adenoma location be uniform throughout the colon, although rectal adenomas are considerably less common than in FAP [68]. Mean age of cancer diagnosis has been reported in the early 50’s [69, 70]. Cumulative risk of CRC in patients with aFAP is estimated to be 69 % by the age of 80 [71].
Clinical Evaluation
The aFAP phenotype occurs in less than 10 % of FAP patients. The clinical criteria [72] for diagnosis is listed in the table (Table 20.4).
Table 20.4
Clinical diagnostic criteria for attenuated FAP (aFAP)
1. No family members with > 100 adenomas diagnosed before the age of 30 and one of two below |
|---|
(a) ≥2 patients with 10–99 adenomas at age >30 years |
(b) One patient with 10–99 adenomas at age ≥30 years and a first-degree relative with CRC |
Pathologically, adenomas may be either pedunculated or sessile and may have tubular, villous, or tubulovillous histology, much like FAP. However, there is a higher chance of sessile polyps that are seen in aFAP patients compared to FAP patients [71]. Extracolonic manifestation, such as desmoids, osteomas, and periampullary tumors, occurs in aFAP patients. CHRPE, however, has not been reported in aFAP patients [73].
Genetics
Mutations in the aFAP patients tend to be at either the 5′ end or 3′ end of APC gene, usually codons 78–167, codons 1581–2843, and in exon 9 [74]. It is hypothesized that the mutations seen in aFAP may result in a weakly functional protein, whereas other APC mutations may cause more severe phenotypes through a complete dysfunctionality of the transcribed protein. Smith et al. reported that 5′ mutations led to unstable proteins that were ultimately degraded, while 3′ mutations resulted in proteins that formed heterodimers which inhibited tumor suppressor function [75].
Surveillance
In patients with a known APC mutation and a family history of AAPC, initial colonoscopy should begin at age 15. If the study shows findings consistent with FAP, then surgery is warranted. If the polyposis is not severe, endoscopic control with regular polypectomies may be feasible and repeated annually. If no polyps are found and the patient is APC mutation positive, colonoscopy annually starting at 20 is recommended. If the APC mutation status is unknown, colonoscopy every 2 years is sufficient. If no polyps are found and the patient tests negative for the APC mutation, routine colorectal cancer screening can be applied. Individuals with a positive family history but negative APC mutation should have a screening colonoscopy at age 15. If no adenomas are found, they can be followed with a colonoscopy every 2 years starting at age 20 [3].
Treatment
Surgical
In patients with mild adenomas, repeated endoscopic polypectomies may be preferable to surgery. In those where the colonic polyps cannot be controlled, prophylactic surgery can be recommended. Most recommend total colectomy with IRA as oppose to IPAA due to rectal sparing [3]. One study reported a 10 % cumulative risk of secondary proctectomy and 3.7 % cumulative risk of rectal cancer following IRA [48].
MYH-Associated Polyposis
MYH-associated polyposis (MAP) is an autosomal recessive disorder due to biallelic mutations (pertaining to both allelles) in MYH, a base excision repair gene. This disease came into light in 2002 when 3 out of 7 siblings presented with multiple adenomatous colorectal polyps and cancer without germline APC mutations [76]. Subsequent APC mutation analysis revealed a result that was characteristic for a defective base excision repair. Unaffected relatives were heterozygous for MYH mutations or wild type, confirming the autosomal recessive pattern of disease inheritance.
The mean age of diagnosis is late 40’s and 50’s, similar to aFAP. Approximately 60 % of MAP patients with polyposis have colorectal cancer initially [77]. Synchronous cancers occur in up to 24 % of patients [78]. The estimated cumulative risk of CRC by age 70 in biallelic MYH mutation carriers has been reported to be as high as 80 % [79]. Penetrance of CRC in MAP patients has been shown to be approximately 19 % at age 50 and 43 % by age 60 [80]. Additionally, there is a twofold increase in risk of CRC for heterozygous carriers of MYH mutations compared to the general population [81].
Genetics
The MUTYH gene, located on chromosome locus 1p34.3–p32.1, is a base excision repair gene that codes for a glycosylase protein [82]. This protein is involved in repairing guanine residues that have undergone oxidative damage. Over 105 mutations have been identified for the MUTYH gene, majority of them being missense mutations. In the western population and in the northern European descent, the most common mutations within the MUTYH gene are the Y179C and the G396D mutations, with reports of approximately 90 % of the MAP patients carrying at least one of these mutations. Different mutations in patients of Indian (E480X), Pakistani (Y90X), southern European (1395 del GGA), and Portuguese (1186–1187 insertion GG) descent have been reported.
Clinical Evaluation
Most patients with biallelic MUTYH mutations present with between 10 and a few hundred polyps. There is a slight propensity for CRC to arise proximal to the splenic flexure [83]. Phenotypic presentation for MAP patients with biallelic G396D mutations was less severe than for Y179C mutation patients [77].
Extracolonic lesions associated with MUTYH mutations include small bowel polyposis, specifically duodenal polyposis, gastric cancer, endometrial cancer, breast cancer, and low to moderate risks in skin, ovarian, and bladder cancers [82]. Very rarely, MAP patients have developed sebaceous gland tumors [84].
Surveillance
Current recommendation [85] is to begin colonoscopic surveillance for MAP patients by age 18–20 to be repeated every 2 years. If polyposis is mild, patients can be followed with polypectomy. When the polyposis becomes severe, surgery is indicated.
Upper gastrointestinal tract screening is advised to begin at the age of 25–30. Recommended screening interval is determined by the Spigelman classification (see FAP section).
Treatment
Polyposis can be controlled with endoscopic polypectomies. Surgical intervention depends on rectum involvement. If rectum is not involved, TAC/IRA is recommended. If rectum is involved, total proctocolectomy with IPAA is recommended. Postsurgically, yearly surveillance with endoscopy is warranted.
Peutz–Jeghers Syndrome
Peutz–Jeghers syndrome (PJS) is an autosomal-dominant disorder with variable penetrance characterized by mucocutaneous melanotic macules and intestinal hamartomatous polyps. Although dysplastic and carcinomatous changes in hamartomas are low (approximately 1 %), malignant transformations are found in PJS patients due to their high polyp burden, especially in the intestines. Incidence of disease varies, ranging from 1 in 8,500 to 1 in 200,000 depending on various reports in the literature [86], showing that the true incidence remains unclear.
Lifetime risk of small bowel cancer risk for patients with PJS has been estimated to be approximately 57 % [87]. Cumulative risk of colonic or extracolonic cancer is 85–93 % by age 70 in PJS patients [87, 88].
Genetics
PJS is caused by a mutation in the LKB1 gene, located on the telomeric region of chromosome 19p13.3. LKB1 is a serine/threonine kinase which complexes with STE-20-related adaptor (STRAD) and mouse protein 25 (MO25) to phosphorylate and mediate downstream cell signaling cascade [89]. It is the only known tumor suppressor kinase. The Human Genome Organization designated the name serine/threonine kinase 11 (STK11) for LKB1. It is the only gene whose mutation is associated with PJS. It can be found in approximately 75 % of PJS patients [86] (Fig. 20.6).
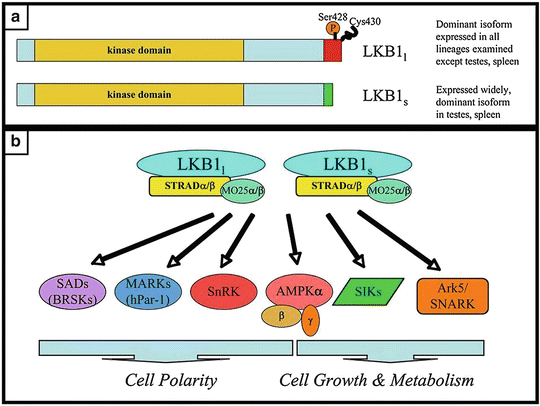
Fig. 20.6
LKB1 is a serine/threonine kinase which complexes with STRAD to phosphorylate downstream kinases in the AMP-activated protein kinase family (AMPK) (Reprinted from Ref. [131]. With permission from Portland Press Limited)
For those without detectable LKB1 mutation, possibilities include large rearrangements of the LKB1 gene due to deletions, duplications or inversions, mutations to the LKB1 promoter, or the existence of additional PJS loci that is yet to be discovered. Thus far, no additional mutations have been found [86].
Gene sequencing is used for genetic screening. Those that come back with a negative result may opt for a multiplex ligation-dependent probe amplification (MLPA). Reported accuracy for genetic testing in at-risk individuals with established family mutations is 95 % [90]. Due to the high false-negative rate, a clinical diagnosis of PJS stands even when the genetic testing is negative. Genetic testing can also be difficult to coordinate due to a limited number of laboratories offering the test and the cost.
Clinical Evaluation
Mucocutaneous melanin pigmentation on or around the lips generally appear by the end of the first year of life and are almost always present by the age of 5 [91]. They can also be seen in the buccal mucosa, periorbital or periaural area, dorsal surface of fingers or toes, and around the anus and genitalia. By puberty and adulthood, these skin pigmentations can disappear so the absence of these lesions in adults does not rule out PJS. They are usually macules 1–5 mm in diameter and vary in color from light brown to black.
Gastrointestinal polyps can occur anywhere in the GI tract, with jejunum being the most common location. Other common locations include the ileum, colon, rectum, stomach, duodenum, appendix, and esophagus [92]. Polyp numbers can vary, from only a handful to thousands.
The majority of PJS patients initially present with small bowel obstruction secondary to intussusception of hamartomas. Most present between the ages of 6 and 18 [93]. Symptoms include abdominal pain, nausea, vomiting, and bloody stool. CT scan is the imaging choice for diagnosis.
According to the guidelines from Mayo Clinic [86], in patients without a family history of PJS, if either of the following two is present, a diagnosis of PJS can be made:
1.
Characteristic mucocutaneous melanotic macules and one or more intestinal polyps with PJS-type histology
2.
Two intestinal polyps with PJS-type histology
In patients with a family history of PJS in a parent or sibling, if any of the following are present, a diagnosis of PJS can be made:
1.
Characteristic melanotic macules
2.
One intestinal polyp with PJS-type histology
3.
An LKB1 mutation
Histologically, the polyps seen in PJS are disorganized hamartomas characterized by hypertrophy or hyperplasia of smooth muscle in the muscularis mucosa. Unique to PJS-type polyps, smooth muscle cells arborize into the superficial epithelial layer. Sometimes the epithelium can invade and be entrapped in the smooth muscle layer, termed pseudo-invasion [86]. This can be mistaken for malignant invasion and can be misdiagnosed as cancer. Therefore, to diagnose a malignancy in PJS polyps, cellular atypia or increased mitotic rate must be seen [94]. In a past review, 10 % of PJS-type polyp specimen examined under microscopy had pseudo-invasion.
Patients with PJS are at increased risk of developing both intestinal and extraintestinal malignancies. One study reported a 15.2 relative risk for PJS patients for all cancers, with statistically increased risk for developing cancer in the esophagus, stomach, small intestines, colon, pancreas, lung, breast, uterus, and ovary [88]. Another study reported an association of PJS and nasal polyposis [91]. Nasopharyngeal carcinoma has also been reported in PJS patients [95]. Gallbladder polyps, gallbladder cancer, and bile duct cancer have also manifested in PJS patients [96–98]. Rarely, hamartomatous polyps have been reported in the ureter [99] and respiratory tract [100] in PJS patients. Finally, several rare cancers have a special association with PJS. In female PJS patients, a highly differentiated adenocarcinoma of the cervix can develop called adenoma malignum (ADM). Special sex cord tumor with annular tubules (SCTAT) can also develop in the ovaries. In males, the corresponding sex cord tumor is the Sertoli cell testicular tumors83.
Surveillance
Riegert-Johnson et al. outlined the surveillance protocol from two institutions, Johns Hopkins Hospital and Mayo Clinic [86]. Both institutions recommend breast self-examination at age 18 with clinical semiannual examination and optional annual mammography annually starting at age 25, endoscopy surveys for stomach and small intestines every 1–8 years starting at age 8, and colonic endoscopic examination every 2–3 years starting at age 18. Johns Hopkins recommends surveillance of pancreas (with endoscopic ultrasound, CT or MR, and/or CA 19–9 as options) and female reproductive organs (ultrasound, serum CA-125, Pap smear) at age 25 and repeated annually, whereas Mayo Clinic recommends these examinations to start at age 18. Clinical examination with ultrasound adjunct for testicular screening should begin at birth and offered annually until age 12.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


