Chapter Outline
ONTOGENY OF THE HUMAN HEMOSTATIC SYSTEM
Natural Inhibitors of Coagulation
The Fetal and Neonatal Fibrinolytic System
Blood Vessel Wall: Age and Anticoagulant Properties
Maternal Preeclampsia and Fetal Hemostasis
Summary of Hemostatic Differences between Neonates and Older Children and Adults
CLINICAL ASPECTS OF DEVELOPMENTAL HEMOSTASIS
Development of the human hemostatic system begins in utero and continues until well after birth. As a result, functional levels of many of the procoagulants, coagulation inhibitors, fibrinolytic components, and platelet-associated factors differ from those of older children and adults, with “adult” values often not reached until about 6 months of age. These unique aspects of fetal and neonatal coagulation were first fully documented in the pioneering work of Dr. Maureen Andrew in the 1980s and have been termed developmental hemostasis . A firm understanding of these differences, as well as access to the “normal” ranges of various hemostatic factors in infants at different gestational and postnatal ages, is critical to proper interpretation of laboratory tests performed on newborns and infants. Despite broad differences from adults in functional levels of hemostatic factors, healthy newborns rarely have difficulties with hemorrhage or thrombosis. Thus these differences in prothrombotic and antithrombotic factors are uniquely balanced in the neonate and fetus and represent a normal physiologic state. However, this system provides little reserve, which often contributes to significant morbidity in sick and preterm infants. In addition to these developmental aspects of hemostasis, many inherited bleeding and clotting disorders can present in the newborn period. This chapter describes the normal development of the hemostatic system and reviews disorders of hemostasis and thrombosis that can occur in the newborn period. Data tables listing normal ranges of hemostasis laboratory tests for term and preterm infants are included in the Appendix. For additional discussion of the investigation and management of neonatal hemostasis and thrombosis, see several recent evidence-based guidelines.
Ontogeny of the Human Hemostatic System
Coagulation Factors
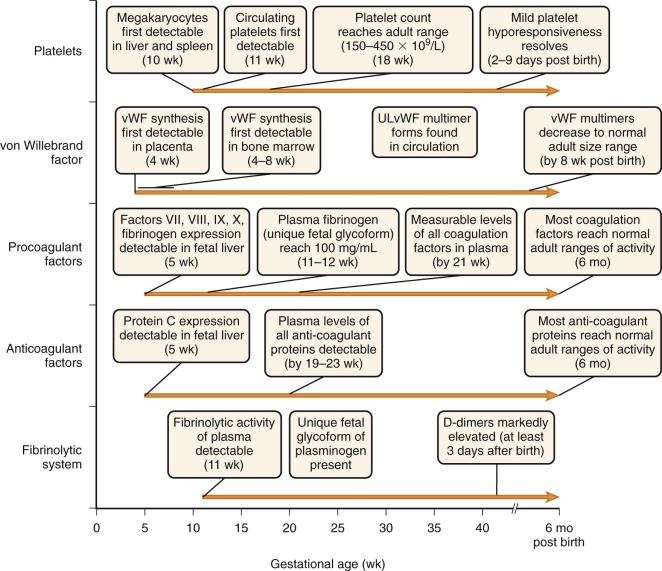
The biochemistry and function of the individual coagulation factors are reviewed in detail in Chapter 27 . Coagulation proteins do not effectively cross the placental barrier and are independently synthesized by the fetus. Messenger RNA (mRNA) transcripts for coagulation factors VII, VIII, IX, and X and fibrinogen are detectable at about 5 weeks of gestation in hepatocytes of the embryo ( Fig. 5-1 ). By 10 weeks of gestation, plasma protein levels of most factors are measurable but range from 10% of the normal adult level for factor IX to about 30% for most of the other coagulation factors. In general, these levels continue to increase gradually in parallel with gestational age ( Appendix Table A5-1 ) . However, with the exception of fibrinogen and factors V, VIII, and XIII, these levels remain considerably below the normal adult range, even at the time of birth for full-term infants. They then continue to gradually increase and reach the normal adult range by about 6 months of age ( Appendix Tables A5-2 and A5-3 ). Premature infants have an accelerated increase in levels after birth and, in general, “catch up” to the full-term infant range by 3 months’ postnatal age ( Appendix Table A5-4 ). True reference ranges for extremely premature infants (<30 weeks’ gestation) are not available, because the majority of these infants have postnatal complications. Appendix Table A5-5 provides reference ranges for older children. The nearly normal “adult” levels of fibrinogen and factors V and VIII in neonates make them useful markers for the investigation of possible consumptive coagulopathy or hemophilia A.
Regulation of fetal coagulation protein levels occurs by both transcriptional and posttranscriptional mechanisms. In addition, ratios of hepatocyte levels to plasma levels of many factors are relatively high in comparison with adults, suggesting that delayed hepatocyte release may also contribute to the relatively low coagulation factor levels in fetuses and newborns.
Qualitative differences in procoagulant proteins also exist between neonates and adults. Fibrinogen from fetuses has increased sialic acid content, increased phosphorus content, and different chromatographic profiles compared with fibrinogen from adults.
Natural Inhibitors of Coagulation
Like many of the procoagulant factors, functional levels of most coagulation inhibitors in fetuses and neonates are significantly lower than those in adults ( Appendix Tables A5-6 and A5-7 ; see also Appendix Table A5-3 ). Awareness of these differences is critical to proper interpretation of laboratory studies obtained during evaluation of neonatal thrombosis. Most coagulation inhibitors reach the adult range of activity by 6 months of age.
As reviewed in Chapter 27 , at least three mechanisms/pathways exist for inhibition of procoagulant activity: (1) cleavage of factors V and VIII by the protein C/S system; (2) direct inhibition of thrombin by antithrombin (AT), heparin cofactor, and α 2 -macroglobulin; and (3) inhibition of factor VIIa by the tissue factor pathway inhibitor (TFPI)/factor Xa complex. When thrombin binds to thrombomodulin on endothelial surfaces, it no longer cleaves fibrinogen but it activates protein C. Activated protein C, in complex with protein S, proteolytically inactivates factors V and VIII. At birth, plasma concentrations of protein C are quite low and remain decreased during the first 6 months of life. Neonates also have about a twofold higher amount of the single-chain form of protein C than adults do, and they may exhibit increased protein C glycosylation. Despite these alterations, there is no evidence that protein C from neonates differs functionally from that of adults when compared at equivalent concentrations.
In adults, protein S circulates in either a free form or in a form that is bound to the carrier protein C4b binding protein. In fetuses and neonates, circulating protein S is almost entirely of the free type because levels of C4b binding protein are extremely low. Thus the protein S activity-to-antigen ratio is greater for fetuses and neonates than for adults. Nonetheless, both total and free protein S levels are lower in fetuses and neonates. They rise gradually after birth, with the adult range of free protein S reached by 4 months and total protein S by about 10 months. The interaction of protein S with activated protein C in the plasma of newborns may be modulated by increased levels of α 2 -macroglobulin.
Healthy full-term newborns have lower functional AT levels than adults do, with an average of 0.55 U/mL based on several studies. This is most likely due to a reduced amount of absolute AT protein inasmuch as most studies have found no significant difference in the antigen-to–functional activity ratio or the immunoelectrophoretic or chromatographic properties of purified AT from neonates and adults. The reduced AT levels in neonates probably account, at least in part, for the relative heparin resistance seen in this age group. AT levels typically reach the adult range by 3 months of age in full-term healthy infants. Preterm infants with respiratory distress syndrome (RDS) have significantly lower levels of AT, typically less than 0.2 U/mL, and the AT present often has additional dysfunctional properties. Lower AT levels correlate with increased catheter-related thromboses, intracranial hemorrhages (ICH), and mortality rates.
Like AT, levels of heparin cofactor II are also substantially lower in infants than adults. A circulating fetal dermatan sulfate proteoglycan has been described that has natural anticoagulant properties mediated through heparin cofactor II. This fetal anticoagulant is also present in the plasma of pregnant women and is produced by the placenta. The length of time that it circulates in newborns is not known; however, it is still present during the first week of life in sick premature infants with RDS.
α 2 -Macroglobulin is a more important inhibitor of thrombin in plasma from newborns than in adults. It compensates, in part, for the low levels of AT and heparin cofactor in newborns, even in the presence of endothelial cell surfaces. However, the overall rate of thrombin inhibition is still lower in newborns than adults.
TFPI binds to factor VIIa/tissue factor complexes in a factor Xa calcium-dependent reaction that results in the inhibition of factor VIIa. After generation of small amounts of thrombin, TFPI prevents further generation of thrombin via tissue factor/factor VIIa. TFPI levels in cord plasma have been reported to be approximately 50% of adult values.
Vitamin K–Dependent Factors
Factors II, VII, IX, and X and proteins C, S, and Z undergo posttranslational γ-carboxylation dependent on vitamin K (VK) of certain glutamic acid residues (see Chapter 27 ). Typically, 9 to 12 glutamic acid residues, often clustered within a 45–amino acid region referred to as the “Gla” domain, are modified. γ-Carboxylation of glutamic acids enhances calcium-dependent phospholipid interactions of the coagulation factors and helps regulate the coagulation cascade. Because of the association of relative VK deficiency and hemorrhagic disease of the newborn (HDN; see detailed description later), development of these VK-dependent factors in utero has been the focus of considerable research. During gestation, a steep gradient of VK concentration is maintained across the placenta, with fetal levels being 10% or less of maternal levels. This gradient is even further enhanced during the third trimester. Consequently, VK stores in newborns are low, as shown by low levels of VK in the cord blood and liver of aborted fetuses and increases in VK-dependent coagulation factor activity following VK administration to neonates. The teleologic explanation for maintenance of low fetal VK levels is not known. However, VK has been shown to promote DNA mutagenesis in vivo. For this reason, several investigators have suggested that maintenance of low fetal VK levels might be a mechanism to prevent chromosomal damage during the rapid cellular proliferation of embryogenesis. In addition to these low VK levels, deficiency of VK reductase activity (a rate-limiting step in the reaction) as a result of immaturity of the hepatocyte may contribute to inefficient γ-carboxylation of coagulation factors. In fact, undercarboxylated prothrombin and protein C have been detected in the cord and peripheral blood of up to 7% and 27% of healthy term infants, respectively, and these levels correlate with gestational age. Even when corrected for this relative VK deficiency, levels of VK-dependent factors are still considerably lower than the normal adult range because the values presented in Appendix Tables A5-1 to A5-4, A5-6, and A5-7 were measured in infants who received VK prophylaxis at birth.
VK-dependent glutamic acid carboxylation also occurs on proteins outside the coagulation system, many of which play roles in development. Such proteins include Gas-6, the ligand for the receptor kinases Sky and Axl; the bone growth–related and calcification-related factors osteocalcin and matrix Gla protein; the urine calcium binding protein nephrocalcin; and PRPG1 and PRPG2, two proline-rich predicted signaling proteins expressed in the central nervous system and endocrine tissues. Thus VK is predicted to have pleiotropic effects on development.
Although VK is maintained at low levels in the fetus, further deficiency of VK during gestation can reduce functional levels of the VK-dependent coagulation factors to an even greater extent. Maternal use of certain anticonvulsant medications, including phenytoin, phenobarbital, valproic acid, and carbamazepine, is associated with an increased risk of VK deficiency in the fetus and neonate. Fetal or neonatal bleeding has been linked to maternal use of these medications on rare occasions. Fetal VK deficiency can be prevented in such cases by antenatal oral administration of 10 mg of VK 1 daily to the mother beginning at 36 weeks’ gestation.
Warfarin is a potent VK antagonist commonly used for long-term anticoagulation. It crosses the placenta and inhibits VK-dependent carboxylation in the fetus. Its use during pregnancy at 6 to 12 weeks of gestation is associated with an embryopathy syndrome consisting of limb deformities, nasal hypoplasia, ophthalmologic malformations, mental retardation, hypotonia, and ear abnormalities. Warfarin exposure during the second or third trimester is associated with fetal wastage and central nervous system anomalies. The developmental defects related to warfarin use during pregnancy are thought to be due to bleeding into the developing limb buds or effects on bone and cartilage growth from inhibition of bone-associated Gla proteins, or both.
The Fetal and Neonatal Fibrinolytic System
The enzyme plasmin degrades polymerized fibrin and is responsible for clot dissolution. It is generated from its precursor plasminogen by two known activators, tissue plasminogen activator (tPA) and urokinase plasminogen activator. These activators, in turn, are inhibited by the plasminogen activator inhibitors PAI-1 and PAI-2. In addition, plasmin itself is inhibited by the circulating blood protein α 2 -antiplasmin. See Chapter 28 for a more detailed description of the fibrinolytic system. In newborns, plasminogen levels are only 50% of adult values, α 2 -antiplasmin levels are 80% of adult values, and plasma concentrations of PAI-1 and tPA are significantly greater than adult values ( Appendix Table A5-8 ). The increased levels of tPA and PAI-1 found on day 1 of life contrast markedly with values obtained from cord blood, which are significantly lower than adult levels. The discrepancy between newborn and cord plasma concentrations of tPA and PAI-1 may be explained by the enhanced release of both proteins from the endothelium shortly after birth. PAI-2 levels are detectable in cord blood but are significantly lower than they are in pregnant women. Unique glycoforms of plasminogen with an increased content of mannose and sialic acid exist in the fetus. This fetal form of plasminogen is less efficiently converted to plasmin by tPA compared with adult plasminogen. No significant differences exist in activation kinetics by urokinase plasminogen activator.
D-Dimers
D-dimers are the breakdown product of fibrin mesh cross-linked by factor XIII. Neonates have markedly elevated D-dimer levels in comparison with older children and adults that persist for at least 3 days after birth ( Appendix Table A5-9 ). These high levels suggest activation of the coagulation system at birth or decreased clearance of D-dimers in neonates, or both.
von Willebrand Factor
von Willebrand factor (VWF) is a large protein that mediates adhesion of platelets to exposed subendothelium and is involved in platelet aggregation (see Chapter 26 ). Its synthesis can first be detected as early as 4 weeks of gestation in placental endothelial cells and by 4 to 8 weeks in bone marrow. In contrast to other coagulation factors, VWF functional levels are considerably elevated in neonates in comparison with the normal adult range (see Appendix Tables A5-2 and A5-5 ). They gradually decline to the normal adult range by about 3 to 6 months. It is important to keep these physiologically elevated levels in mind when using VWF as a marker of inflammation in infants.
VWF is released from endothelial cells and megakaryocytes as large multimers with molecular weights in the millions-of-dalton range. They are then cleaved into smaller multimers by the metalloproteinase ADAMTS-13. Unusually large VWF (ULVWF) multimers are VWF multimer forms larger than those found in normal plasma. These ULVWF multimers are 10 to 20 times more active in shear stress–induced platelet aggregation and bind more avidly to the extracellular matrix of fibroblasts than do the VWF multimer forms found in normal plasma. In older children and adults, the presence of ULVWF plasma forms is the hallmark of thrombotic thrombocytopenic purpura, a microangiopathic disorder. It is due to deficiency, acquired or congenital, of ADAMTS-13. ULVWF forms are consistently found in platelet-poor plasma (PPP) from fetuses less than 35 weeks’ estimated gestational age (EGA) and most fetuses older than 35 weeks’ EGA, as well as in umbilical cord blood. These forms are not seen in simultaneously sampled maternal PPP, indicating that the ULVWF forms are fetal specific. The ULVWF in fetal PPP is similar to the VWF directly released from endothelial cells. This similarity may be explained by the finding of low VWF cleaving protease (ADAMTS-13) activity in many neonates. Serial monitoring of PPP from neonates shows a gradual reduction in ULVWF to the normal size range within 8 weeks after birth. The presence of ULVWF during fetal development is probably physiologically relevant because plasma derived from umbilical cord blood shows increased shear stress–induced and ristocetin-induced VWF platelet-binding activity in comparison with plasma derived from adults, even when adult platelets are used in the assay. The enhanced hemostatic efficacy of ULVWF may therefore partially balance the low functional levels of other coagulation factors to achieve reliable hemostasis in the fetus and newborn.
Platelets
Megakaryocytes During Fetal Development
In humans, megakaryocytes are first detectable in the liver and spleen by 10 weeks of gestation. By 30 weeks, megakaryocytes are present in bone marrow and actively contribute to thrombopoiesis. Circulating megakaryocyte progenitors are detectable in the blood of neonates, and their number decreases as a function of postconceptional age (gestational age plus days of life). Neonatal megakaryocytes have distinct properties compared with adult megakaryocytes, including increased thrombopoietin sensitivity, increased proliferation, lower ploidy, and increased cytoplasmic maturation for the degree of ploidy. The differences in megakaryocyte size resolve by about 4 years of life.
Platelet Number, Size, and Survival During Development
In humans, circulating platelets are first detected by 11 weeks’ gestational age. Thereafter, the plasma platelet concentration rises rapidly and reaches the adult range of 150 to 450 × 10 9 /L by about 18 weeks of gestation. During the earliest phase of platelet production, mean platelet volume is larger than the adult range. However, platelet volume “normalizes” soon thereafter and reaches the adult range of 7 to 9 fL. It is possible that the larger platelets arise from a unique wave of “primitive” megakaryopoiesis that occurs early in gestation, as demonstrated in several mouse studies, but this mechanism remains to be determined in humans. Platelet survival has not been measured in healthy infants. In rabbits, survival of 111 In-oxine–labeled platelets is similar in adult and newborn rabbits. However, in humans, levels of reticulated platelets, which represent those recently released from megakaryocytes, are elevated about twentyfold in comparison with adults. Concentrations of thrombopoietin (TPO), the major cytokine for megakaryocyte progenitor proliferation and development, are increased about twofold in fetal versus adult plasma. These findings suggest possible increased platelet turnover in human fetuses versus adults.
Platelet Structure
Peripheral blood platelets from newborns contain similar numbers of platelet-specific granules as platelets from older children and adults; however, serotonin and adenosine diphosphate (ADP), which are stored in dense granules, are present at concentrations of less than 50% of adult values. Newborn platelets contain normal levels of the receptors glycoprotein Ib (GPIb) (part of the VWF binding complex), GPIa/IIa, P-selectin, and HPA-1a. Variable levels of GPIIb/IIIa have been reported, with some studies indicating significantly reduced expression. Functional epinephrine receptors are significantly diminished in newborn platelets.
Platelet Function
Platelet aggregometry and other functional assays of neonatal and cord blood platelets have shown variable results, but most demonstrate a modest hyporesponsiveness to ADP, epinephrine, collagen, and thrombin when compared with platelets from older children and adults ( Fig. 5-2 ). The hyporesponsiveness spontaneously resolves by 2 to 9 days after birth in full-term infants. Extremely low birth weight neonates (<1 kg, gestation < 30 weeks) have markedly hyporesponsive platelets. This impaired platelet response may contribute to the propensity of extremely low birth weight neonates for ICH. The platelet hyporesponsiveness improves over the first 10 to 14 days after birth, but platelet reactivity does not reach adult levels even by day 14. Several explanations have been proposed to account for this hyporesponsiveness, including impaired calcium channel transport and signal transduction, decreased availability of α-adrenergic receptors because of either delayed receptor maturation or occupation by catecholamines released during birth, and the presence of inhibitory placenta-derived prostaglandins.

In contrast to the agonists just described, agglutination in response to low concentrations of ristocetin is significantly enhanced in fetal and newborn platelet-rich plasma in comparison with that of adults, probably partly because of high VWF levels and the presence of ULVWF forms in newborn plasma. In the end, the hyporesponsiveness to certain platelet agonists and the hyperresponsiveness to VWF appear to balance each other out inasmuch as most healthy term infants do not experience bleeding or thrombotic complications in the newborn period.
Platelet Activation During the Birth Process
There is strong evidence that platelets are activated during the birth process. Cord plasma levels of thromboxane B 2 , β-thromboglobulin, and platelet factor 4 are increased; the granular content of cord platelets is decreased; and epinephrine receptor availability is reduced, perhaps as a result of occupation. The mechanisms of activation are probably multifactorial and include thermal changes, hypoxia, acidosis, adrenergic stimulation, and the thrombogenic effects of amniotic fluid.
Blood Vessel Wall: Age and Anticoagulant Properties
The endothelium fulfills a complex role in hemostasis in that it prevents thrombotic complications under physiologic conditions and promotes fibrin formation when injured. One of the anticoagulant properties of endothelial cell surfaces is mediated by lipoxygenase and cyclooxygenase metabolites of unsaturated fatty acids. Prostacyclin (prostaglandin I 2 ) production by cord vessels exceeds that by blood vessels in adults. A second endothelial cell–mediated antithrombotic property is promotion of AT neutralization of thrombin by cell surface proteoglycans. Structurally, there is evidence that the vessel wall glycosaminoglycans of the young differ from those of adults. In a rabbit venous model, increased amounts of glycosaminoglycans are seen in the inferior venae cavae of rabbit pups as compared with adults. Similarly, in the rabbit arterial model, greater glycosaminoglycan-mediated AT activity is seen in rabbit pups than in adult rabbits.
Maternal Preeclampsia and Fetal Hemostasis
Preeclampsia is a pregnancy-associated syndrome consisting of maternal hypertension and proteinuria. Alterations in maternal coagulation and fibrinolysis, including reduced AT and PAI-2 levels and increased tPA antigen, PAI-1 antigen, and fibrin degradation product levels, have been described in women with preterm preeclampsia. This suggests a state of enhanced thrombin generation and fibrinolysis in the maternal circulation. Analysis of cord blood from infants born to mothers with preeclampsia shows no statistical difference in the levels of these factors versus those of age-matched controls born to mothers without preeclampsia, thus indicating a probable protective effect of the placenta on the fetal circulation.
Summary of Hemostatic Differences Between Neonates and Older Children and Adults
In summary, the salient differences in the hemostatic system of neonates and older children or adults are (1) decreased plasma concentrations of many of the procoagulant proteins, including factors II, VII, IX, X, XI, and XII, prekallikrein, and high–molecular-weight kininogens; (2) a unique fetal glycoform of fibrinogen; (3) decreased plasma concentrations of the coagulation inhibitors AT, heparin cofactor II, TFPI, protein C, and protein S, with a concomitantly slower rate of thrombin inhibition; (4) a unique glycoform of plasminogen that is less efficiently converted to plasmin by tPA; (5) markedly elevated D-dimer levels until at least 3 days after birth; (6) increased plasma VWF concentrations and elevated levels of circulating ULVWF multimers; (7) smaller and more proliferative megakaryocytes; and (8) modest, transient hyporesponsiveness of platelets to certain agonists such as collagen and epinephrine, but increased agglutination with low-dose ristocetin. In general, most of these differences resolve within the first 6 months of life.
Clinical Aspects of Developmental Hemostasis
Hemorrhagic Disorders in Neonates
Although acquired disorders are more commonly seen, severe forms of congenital factor deficiencies or platelet disorders can present in early infancy and should be seriously considered in otherwise healthy infants who are bleeding. About 15% to 30% of children with inherited bleeding disorders have hemorrhage in the neonatal period. In addition, a third of new cases of severe hemophilia represent new mutations and are therefore not accompanied by any antecedent family history.
Clinical Findings
The clinical manifestations of bleeding disorders are different in newborns than in older children and adults. Bleeding may appear as oozing from the umbilicus, bleeding into the scalp, large cephalhematomas, bleeding after circumcision, bleeding from peripheral phlebotomy sites, and bleeding into the skin. A small but important proportion of infants are seen with ICH as the first manifestation of their bleeding tendency. Sick infants can bleed from mucous membranes, the bladder, and sites of invasive procedures. Joint bleeding is rare. The most common causes of bleeding in healthy infants are thrombocytopenia secondary to transplacental passage of a maternal antiplatelet antibody, sepsis, VK deficiency, and congenital coagulation factor deficiencies.
Laboratory Evaluation
In addition to a workup for sepsis, laboratory evaluation of infants with bleeding complications should include determination of the prothrombin time (PT), activated partial thromboplastin time (APTT), thrombin clotting time (TCT), fibrinogen level, and platelet count. As discussed earlier, results need to be compared with the expected range of values for neonates because some may not reflect the normal adult range. It is also critical to apply age-specific reference ranges generated using the same analyzer and reagents because these can influence the measured absolute levels. Tests that provide exogenous AT should be avoided because they do not reflect the normal physiologic state of low AT levels in neonates. Values obtained from cord blood may be different from those obtained from neonatal peripheral blood samples. Abnormalities in test results usually prompt additional tests, such as specific factor assays. For a male child in whom hemophilia A or B is suspected, specific factor assays should be performed regardless of the APTT value. The PT, APTT, and TCT are not prolonged in those with factor XIII and α 2 -antiplasmin deficiency. Therefore levels of these factors must be measured directly if deficiencies are suspected.
Sample Collection.
Collection of samples for laboratory investigation of hemostatic defects in neonates, especially premature infants, presents particular challenges because of the patient’s small size and increased hematocrit. Specialized microcollection tubes (1 mL) should be available in the neonatal unit. Blood sampling techniques should avoid contamination with intravenous fluids and heparin and should also avoid activating the coagulation system. Slow transit time and contact with plastic tubing are most likely to cause activation. All neonatal samples should be inspected for the presence of fibrin clumps before processing, and those containing clumps should not be used for analysis. Ideally, the volume of anticoagulant in the sample should be based on the volume of plasma, not the total volume of blood, because the increased hematocrit of neonates causes accentuated dilution of the coagulation factors if not taken into account. This is particularly important for neonates with very high hematocrit values, such as those with cyanotic congenital cardiac disease. Overfilled or underfilled samples should not be analyzed.
Screening Coagulation Tests in Neonates.
Screening tests of plasma clotting activity, including the PT, APTT, and TCT, are all prolonged during fetal development and in neonates when compared with the normal adult range (see Appendix Tables A5-1 to A5-3 ), probably because of physiologic “deficiencies” of the VK-dependent factors and contact factors and the presence of fetal glycoforms of fibrinogen. The absolute values for these tests, as well as specific coagulation factor tests, depend on the reagents and analyzer used. Appendix Table A5-3 provides reference ranges using the STA Compact analyzer and Diagnostica Stago reagents, a system currently used in many clinical laboratories. In contrast to some earlier systems, such as the ACL system, the STA Compact analyzer is not affected by plasma bilirubin levels, which are frequently elevated in neonates. Reference ranges using the Behring Coagulation System (BCS) and the Sysmex CA-1500 System with various reagents have also recently been published. The differences observed underscore the need for each clinical laboratory to establish its own normal age-related reference ranges.
Bleeding Time.
The bleeding time has traditionally been used as an in vivo test of platelet function, particularly for testing adhesion to injured vasculature. However, it is subject to considerable operator-dependent variability and is not routinely used in newborns. Automated devices modified for newborns and children are available and have been standardized. In contrast to the prolonged coagulation screening tests, multiple studies have shown that bleeding times in infants during the first week of life are significantly shorter than those in adults. This somewhat paradoxic finding can be explained by several mechanisms, including higher plasma concentrations of VWF, enhanced function of VWF because of a disproportional increase in ULVWF multimers, large red cells, and high hematocrits.
Platelet Function Analyzer.
The Platelet Function Analyzer 100 (PFA-100) system provides an in vitro method of assessing primary platelet-related hemostasis by measuring the time (closure time) required for a platelet plug to occlude a microscopic aperture cut into a membrane coated with collagen and either epinephrine or ADP. It uses the patient’s own platelets and plasma. The PFA-100 system is attractive for use in neonates because of the small volume required, rapidity of testing, and standardization of testing cartridges. Like the bleeding time, most studies of neonates’ peripheral or cord blood report PFA-100 closure times, for both epinephrine and ADP, that are shorter than those for older children and adults ( Appendix Table A5-10 ). This is probably due to similar mechanisms as described earlier for the shortened bleeding time. Ranges for premature infants have been reported and show an inverse relationship between ADP closure time and gestational age. Although the PFA-100 system may eventually become a standard screening test for platelet function in neonates, older children, and adults, its clinical utility has not yet been clearly established. Thus further studies will be required to determine the optimal method of assessing primary hemostasis in newborns and children, particularly premature infants.
Hereditary Coagulation Factor Deficiencies
For most hemostatic components, both severe and mild forms of deficiency can occur, with severe deficiencies often characterized by significant bleeding in newborns. Chapters 31 and 32 discuss hereditary factor deficiencies in detail.
Inheritance.
Deficiencies of factors II, V, VII, XI, and XII, prekallikrein, and high–molecular-weight kininogen are rare autosomally inherited disorders, with consanguinity present in many families. Deficiencies of factor XII, prekallikrein, and high–molecular-weight kininogen do not result in hemorrhagic complications and thus are not considered further here. Deficiencies of factors VIII and IX are sex linked and the most common congenital bleeding disorders to present in newborns. Rarely, combined deficiencies of factors II, VII, and IX and/or factors V and VIII may also present in the neonatal period. Tests are available to diagnose most congenital factor deficiencies prenatally so that either termination of pregnancy or management of affected infants can be planned (see Chapters 31 and 32 ).
Clinical Findings.
In the majority of newborns with congenital coagulation factor deficiencies, bleeding does not occur in the perinatal period unless a hemostatic challenge is present. On the other hand, unexplained bleeding in an otherwise healthy newborn should be carefully investigated because it may be due to a congenital coagulation factor deficiency.
ICH is rare in full-term newborns and usually occurs either spontaneously or after insults such as birth asphyxia or trauma, VK deficiency, or severe congenital factor deficiencies. Other risk factors include low birth weight, young gestational age, and ethnic background. The risk for ICH in children with severe hemophilia A or B ranges from 2% to 8%. The location of the ICH is most commonly the subarachnoid area, but subdural and parenchymal bleeding also occurs. Head ultrasound may not detect small parafalcine or subdural bleeding, so computed tomography or magnetic resonance imaging is the preferred modality when deficiency of a coagulation factor is suspected. Some infants require surgical intervention, and many have long-term neurologic deficits.
Full-term infants with unexplained ICH should be carefully evaluated for congenital or acquired hemostatic defects. Unfortunately, the diagnosis of ICH may be delayed because of the nonspecific nature of the early clinical findings, including poor feeding, lethargy, apnea, vomiting, and irritability. Further delays can result when secondary coagulopathies such as disseminated intravascular coagulation (DIC) occur or because of confusion related to the physiologically low levels of some coagulation factors in newborns. The more extreme clinical manifestations of ICH are usually recognized early and include seizures, meningismus, and a tense fontanelle.
Although severe deficiency of factor VIII is the most common cause of ICH from a coagulation factor deficiency, severe congenital deficiencies of fibrinogen and factors II, V, VII, VIII, IX, X, XI, and XIII can also cause ICH at birth. The incidence of ICH in newborns is unknown and is probably changing because of improvements in perinatal care. The widespread use of ultrasound, a safe modality for monitoring fetuses at risk, has resulted in the detection of ICH in utero. In utero factor replacement has been accomplished in several infants. Though less common than ICH, subgaleal bleeding with concurrent shock and DIC may be the initial manifestation of a congenital factor deficiency.
Diagnosis.
In newborns, the diagnosis of many congenital factor deficiencies based on plasma concentrations can be difficult because of neonates’ physiologically low levels at birth (see Appendix Tables 5-2 and 5-3 ). Mild to moderate hereditary deficiencies of factors II, VII, IX, X, and XI result in plasma concentrations that may overlap with the normal neonatal range. In contrast, plasma concentrations resulting from either mild to moderate factor VIII deficiency or severe deficiency of factors V, VII, VIII, IX, X, and XIII can easily be distinguished from physiologic values. Prenatal diagnosis of most hereditary factor deficiencies is performed by amniocentesis or chorionic villus biopsy and is largely confined to severe hemophilia A and B, although deficiencies of factors V, VII, and XIII and VWF have also been diagnosed prenatally.
Treatment.
In the presence of active bleeding or a planned hemostatic challenge, the fundamental principle of management is to increase the plasma concentration of the deficient coagulation protein to a minimal hemostatic level. This target level varies, depending on the protein and nature of the hemostatic challenge (see Chapters 31 and 32 ). When possible, recombinant protein products or detergent-treated pooled donor preparations should be used to reduce the risk of infectious complications. Most products available for the treatment of older children and adults can be used in neonates. Although it has not been well studied, the half-lives and volumes of distribution of recombinant factor VIII (rFVIII) and recombinant factor IX (rFIX) appear to be similar in full-term newborns and older children, so standard initial dosing regimens can be used. However, it is critical to monitor trough levels closely and make appropriate dose modifications to ensure achievement of the proper target level. There are a few case reports of extremely premature infants (<1500 g or <30 weeks’ EGA) having a slightly shortened half-life of factor VIII (about 6 hours vs. 8 to 12 hours in full-term infants and older children).
Fresh frozen plasma (FFP) should be given for bleeding emergencies in neonates in whom a coagulation defect is suspected but a specific factor deficiency has not yet been documented. There are case reports of a small number of infants (<4 months) treated with recombinant factor VIIa (rFVIIa) for catastrophic bleeding of varying causes. However, rFVIIa treatment of patients without congenital factor VII deficiency or hemophilia with high-titer inhibitors should be considered investigational at this time. Infants with known or suspected deficiencies in coagulation factors should receive vaccinations by the subcutaneous rather than the intramuscular route, and arterial puncture should be avoided when possible.
Mode of Delivery.
The optimal mode of delivery for infants of hemophilia carrier mothers has yet to be determined. However, normal vaginal delivery appears to be generally safe for many babies with hemophilia. Use of vacuum assistance is associated with an increased risk for significant cranial hematomas in newborns with hemophilia and should be avoided if possible.
Specific Coagulation Factor Deficiencies
Fibrinogen Deficiency.
Deficiency of fibrinogen is rare. Bleeding secondary to afibrinogenemia has been reported in newborns after circumcision or as umbilical stump bleeding and soft tissue hemorrhage, with some cases being fatal. Reported replacement therapies have included whole blood, cryoprecipitate, FFP, and fibrinogen concentrates. One fibrinogen concentrate (Haemocomplettan HS, Centeon/Aventis Behring) is available in Europe and North America.
Factor II Deficiency.
Deficiency of prothrombin is very rare. Reported bleeding complications in newborns include gastrointestinal bleeding and ICH. Reviews of adult patients have reported bleeding after invasive events such as circumcision and venipuncture or as soft tissue hematomas. Although FFP can be used as initial therapy, factor II concentrate or prothrombin complex concentrate (PCC) is the preferred replacement product.
Factor V Deficiency.
Bleeding as a result of severe factor V deficiency has been reported in newborns. Clinical manifestations include ICH, subdural hematoma, bleeding from the umbilical stump, gastric hemorrhage, and soft tissue hemorrhage. Replacement therapy includes whole blood and FFP. Although thrombotic complications occur in some patients with factor V deficiency, they have not been reported in newborns.
Factor VII Deficiency.
Severe factor VII deficiency (factor VII level <1%) usually causes significant bleeding equivalent to that seen in patients with severe hemophilia. Patients with factor VII levels greater than 5% generally have mild hemorrhagic episodes. The most commonly reported bleeding complication in newborns with congenital factor VII deficiency is ICH. In a review of 75 patients with factor VII deficiency, ICH was observed in 12 (16%). In 5 (42%) of these 12 patients, ICH occurred in the first week of life, with a fatal outcome. Congenital factor VII deficiency may occur in infants with Dubin-Johnson syndrome or Gilbert syndrome in certain populations. Bleeding episodes secondary to factor VII deficiency can be treated with FFP, PCC, purified factor VIIa concentrates, or, preferably, rFVIIa. Lower doses of rFVIIa (15 to 20 µg/kg) than those used for patients with severe hemophilia and high-titer inhibitors (90 µg/kg) are often sufficient to control bleeding in patients with factor VII deficiency. However, this has not been carefully defined in prospective clinical trials. Moreover, dosing has not been optimized for neonates and infants. Caution should be used in combining treatment with rFVIIa and PPC because thrombotic complications have been reported.
Factor VIII Deficiency.
The severity of factor VIII deficiency is determined by the plasma concentration of factor VIII, with a level of less than 1% being severe, 1% to 5% being moderate, and 5% to 50% being mild. Severe factor VIII deficiency is the most common inherited bleeding disorder of the neonatal period. In addition, a small number of neonates with moderate and mild hemophilia are seen after a hemostatic challenge. Large cohort studies have revealed that approximately 10% of children with hemophilia have clinical symptoms in the neonatal period. In this group of factor VIII–deficient children experiencing bleeding in the neonatal period, approximately 50% of bleeding episodes occur after circumcision, with almost 20% of patients suffering intracranial bleeding; some deaths also occur. In contrast to older infants and children, bleeding into joints is extremely rare in neonates. Severe factor VIII deficiency can, on rare occasion, occur in female infants as a result of homozygosity or compound heterozygosity for an FVIII gene mutation or skewed lyonization. Management of bleeding secondary to factor VIII deficiency is discussed in detail in Chapter 31 .
Factor IX Deficiency.
Classification of the severity of factor IX deficiency is identical to that used for factor VIII deficiency. Diagnosis of milder forms of factor IX deficiency is complicated by physiologic levels of factor IX that can be as low as 0.15 U/mL and, in rare infants, by the potential for concurrent VK deficiency. Bleeding after circumcision and ICH can occur in neonates with factor IX deficiency. Management of bleeding secondary to factor IX deficiency is discussed in detail in Chapter 31 .
Factor X Deficiency.
Severe factor X deficiency can be manifested as bleeding in the newborn period. ICH was present in a high proportion of reported cases, several of which were fatal. Additional sites of bleeding that have been noted include umbilical, gastrointestinal, and intraabdominal sites. Whole blood, FFP, factor X concentrate, and PCC have been used as replacement therapy.
Factor XI Deficiency.
Severe deficiency of factor XI is rare. It is different from other coagulation protein deficiencies in that bleeding symptoms are not necessarily tightly correlated with factor plasma concentrations. Bleeding complications as a result of severe factor XI deficiency were reported in two newborns. One bled after circumcision at the age of 3 days and had a factor XI level of 0.07 U/mL. In another newborn, factor XI deficiency with bilateral subdural hemorrhage was diagnosed prenatally. Either FFP or cryoprecipitate can be used for the treatment of factor XI–deficient patients if factor XI concentrate is not available.
Factor XIII Deficiency.
Severe factor XIII deficiency is typically manifested at birth as bleeding from the umbilical stump or ICH. Other clinical manifestations of homozygous factor XIII deficiency include delayed wound healing, abnormal scar formation, and recurrent soft tissue hemorrhage with a tendency to form hemorrhagic cysts. ICH occurs even in the absence of trauma in approximately a third of all affected patients. Newborns with heterozygous factor XIII deficiency are not clinically affected.
FFP, cryoprecipitate, or factor XIII concentrates can be used for the treatment of factor XIII–deficient patients. Newborns with known factor XIII deficiency should receive a prophylactic regimen of factor XIII because of the high incidence of ICH. Plasma concentrations of factor XIII greater than 1% are effective, and the very long half-life of factor XIII permits once-per-month therapy. Therefore prophylactic replacement therapy consists of either small doses of FFP (2 to 3 mL/kg) administered every 4 to 6 weeks, cryoprecipitate at a dose of 1 bag/10 to 20 kg of weight every 3 to 6 weeks, or, preferably, factor XIII concentrate at a dose of 10 to 20 U/kg every 4 to 6 weeks, depending on the clinical situation and the preinfusion plasma concentration of factor XIII.
Hereditary Deficiencies of Multiple Coagulation Factors.
Hereditary deficiencies of two or more coagulation proteins have been reported for 16 different combinations of coagulation factors. Combined deficiency of factors V and VIII has been described in patients with mutations in the genes encoding LMAN1 (also called ERGIC-53) and MCFD2. Administration of FFP is usually the initial therapy. Subsequent treatment varies, depending on the specific factors affected.
Hemorrhagic Disease of the Newborn
Historical Background.
HDN, as first described by Townsend in 1894, consists of hemorrhage on days 1 through 5 of life from multiple sites in otherwise healthy infants. Subsequently, a causal link between HDN and abnormal blood coagulation was established. Initial treatment of infants with HDN consisted of intravenous, intramuscular, or subcutaneous injection of blood or serum. Even at this very early time, the difficulty of separating treatment response from spontaneous improvement was recognized. Later, a randomized, controlled trial showed that intramuscular injection of blood was not helpful in preventing the abnormalities in blood coagulation that occurred during the first week of life. The link between VK deficiency and spontaneous hemorrhage was first recognized in chicks in 1929. The association between VK deficiency and HDN quickly followed, as subsequently did treatment of infants with HDN.
The next historical step was recognition of the link between decreased prothrombin activity and increased PT on days 2 to 4 of life in the absence of prophylactic VK. Prothrombin activity was observed to return to normal by days 5 to 7 of life. These observations led to the hypothesis that VK administered prophylactically could prevent HDN. There was uniform agreement that the prophylactic administration of VK to mothers or infants prevented the decrease in prothrombin activity during the first 3 to 4 days of life. On the basis of these studies, VK prophylaxis was widely recommended. Subsequently, the scientific basis for this policy became less clear for several reasons. First, plasma concentrations of other coagulation proteins in addition to prothrombin were shown to be low in newborns. Second, there was increasing recognition that bleeding in neonates was often not due to VK deficiency. Third, administration of high amounts (50 to 70 mg) of a water-soluble form of VK resulted in hemolytic anemia with kernicterus in some infants. Fourth, many clinicians suggested that VK prophylaxis was not needed for all healthy full-term infants. Consequently, VK prophylaxis was suspended in some countries, and recurrence of HDN ensued. Most of the controversy concerning prophylactic use of VK can be explained by the design of the trials and subsequent interpretation of their results. The best evidence comes from large randomized, placebo-controlled trials. Such trials have consistently shown a statistically significant benefit from VK prophylaxis in terms of clinical bleeding. No randomized, controlled trials with large enough sample size have shown that prophylactic VK does not prevent bleeding.
Prophylactic Vitamin K Administration.
VK exists in three forms: VK 1 (phytonadione), which is present in leafy green vegetables; VK 2 (menaquinone), which is synthesized by intestinal bacterial flora; and VK 3 (menadione), which is a synthetic, water-soluble form. VK 3 is rarely used in newborns because at high doses it causes hemolytic anemia, which results in jaundice and potential morbidity. The recommendations for VK prophylaxis in many countries are similar. Daily requirements of VK are approximately 1 to 5 µg/kg of body weight for newborns. Most groups recommend a single dose of 0.5 to 1 mg intramuscularly or an oral dose of 2 to 4 mg at birth, with subsequent dosing for breastfed infants. Oral VK prophylaxis is preferable to parenteral prophylaxis; studies have shown that oral administration of VK is as effective, less expensive, and less traumatic than intramuscular administration in preventing the classic signs and symptoms of VK deficiency. However, orally administered VK 1 or VK 3 may not be as effective as intramuscularly injected VK in the prevention of late VK deficiency. Strategies for preventing late VK deficiency include repeat administration of oral VK or continuous low-dose VK supplementation. A mixed micelle oral VK 1 preparation that is readily absorbed has been tested in children. However, optimal dosing of this preparation to prevent late VK deficiency bleeding remains to be determined.
In addition to general prophylaxis at birth, patients in certain risk groups require additional VK prophylaxis (e.g., infants with α 1 -antitrypsin deficiency, chronic diarrhea, cystic fibrosis, or celiac disease). Pregnant women receiving oral anticonvulsant therapy should take 10 mg of oral VK 1 daily beginning at 36 weeks’ gestation for the prevention of overt VK deficiency in their infants at birth.
In 1990, a cohort study aimed at determining perinatal risk factors for childhood cancer reported an association between drugs administered in the peripartum period (maternal pethidine or neonatal VK) and childhood cancer (odds ratio, 2.6). A subsequent case-control study by the same group found no association between maternal pethidine and childhood cancer; however, they again reported an increased risk of childhood cancer after neonatal intramuscular VK administration (odds ratio, 1.97). In contrast, a later large case-control study of 2530 children with cancer, one ecologic study, and one metaanalysis have shown no association between VK administration and childhood cancer. The current consensus of the American Academy of Pediatrics Committee on Fetus and Newborn is that the risk for increased childhood cancer is probably minimal, if any, and the benefits of VK prophylaxis in terms of reduced bleeding are substantial.
Bleeding Caused by Vitamin K Deficiency in the Newborn.
In the absence of prophylactic VK, the incidence of vitamin K–dependent bleeding (VKDB) ranges from 0.25% to 1.7%. Infants are at greater risk for hemorrhagic complications from VK deficiency than similarly affected adults are because their plasma concentrations of VK-dependent factors are physiologically decreased. The clinical manifestation of VKDB can be classified as classic, early, or late on the basis of the timing and type of complications ( Table 5-1 ). Classic VKDB initially occurs on days 2 to 7 of life in breastfed, healthy full-term infants. Causes include poor placental transfer of VK, marginal VK content in breastmilk (<20 mg/L), inadequate milk intake, and a sterile gut. VKDB rarely occurs in formula-fed infants because commercially available formulas are supplemented with VK. How often classic VKDB occurs in the absence of VK prophylaxis depends on the population studied, the supplemental formula, and the number of mothers breastfeeding. Early VKDB develops in the first 24 hours of life and is linked to maternal use of specific medications that interfere with VK stores or function, such as some anticonvulsants. Late VKDB occurs between weeks 2 and 8 of life and is linked to disorders that compromise ongoing VK supply.
| Parameter | Early Form | Classic Form | Late Form |
|---|---|---|---|
| Age | <24 hours | 2-7 days | 0.5-6 months |
| Causes and risk factors | Medications during pregnancy Anticonvulsants Oral anticoagulants (rifampin, isoniazid) Antibiotics (rarely, idiopathic or hereditary) | Breastfeeding Inadequate VK intake | Marginal VK content in breastmilk resulting from low VK intake and absorption Cystic fibrosis Diarrhea α 1 -Antitrypsin deficiency Hepatitis Celiac disease |
| Localization in order of occurrence | ICH Gastrointestinal region Umbilicus Intraabdominal region Cephalhematoma | ICH Gastrointestinal region Umbilicus ENT region Injection sites Circumcision | ICH (>50%) Gastrointestinal region Skin ENT region Injection sites Urogenital tract Intrathoracic region |
| Occurrence without VK prophylaxis | Very rare | 1.5% (1/10,000 births) | 4-10/10,000 births * |
| Prophylaxis | Discontinue or replace offending medications Maternal VK prophylaxis | Adequate VK supply Early and adequate breastfeeding Formula VK prophylaxis | Adequate VK supply Adequate breastfeeding Formula VK prophylaxis † |
* More common in Southeast Asia.
† A single intramuscular injection is better than a single oral dose; repeated small doses are closer to physiologic conditions.
Laboratory Diagnosis of Vitamin K Deficiency in the Newborn.
Laboratory tests used to detect VK deficiency include screening coagulation tests, specific factor assays, measurement of decarboxylated forms of VK-dependent factors, PIVKA (protein induced by VK antagonists) assays, and direct measurements of VK levels. The results of these tests must always be compared with values for age-matched, healthy, non–VK-deficient infants to distinguish physiologic and pathologic deficiencies.
Treatment.
An infant suspected of having VK deficiency should be treated immediately with VK while laboratory confirmation is awaited. All infants with VKDB should be given VK either subcutaneously or intravenously, depending on the clinical problem. VK should not be given intramuscularly to infants with VKDB, because large hematomas may form at the site of the injection. Absorption of subcutaneously administered VK is rapid, and its effect occurs only slightly slower than that of systemically administered VK. Intravenous VK should be given slowly and with a test dose because it may induce an anaphylactoid reaction. Infants with major bleeding because of VK deficiency should also be treated with plasma products to rapidly increase levels of VK-dependent proteins. Plasma is the product of choice for treatment of a non–life-threatening hemorrhagic event, whereas PCCs should be considered for the management of life-threatening bleeding.
Liver Disease
The coagulopathies associated with liver disease in newborns are similar to those of adults and reflect the failure of hepatic synthetic function. However, in newborns this problem may be superimposed on the already “physiologic” immaturity of the coagulation system, activation of the coagulation and fibrinolytic systems, poor clearance of activated coagulation factors, and loss of hemostatic proteins into ascitic fluid. Secondary effects of liver disease on platelet number and function also occur in newborns. Common causes of hepatic dysfunction in newborns include viral hepatitis, hypoxia, total parenteral nutrition, shock, and fetal hydrops. Rare causes include genetic diseases such as α 1 -antitrypsin deficiency, galactosemia, and tyrosinemia.
Laboratory abnormalities induced by acute liver disease include prolongation of the PT and low plasma concentrations of several coagulation proteins, including fibrinogen. However, it is important to compare these levels with those of normal age-matched controls (see Appendix Tables A5-1 to A5-3 ). Fibrinogen is present at adult levels in newborns and may be a useful marker. Chronic liver failure with cirrhosis is also characterized by a coagulopathy and mild thrombocytopenia. Secondary VK deficiency may occur as a result of impaired absorption from the small intestine, particularly in those with intrahepatic and extrahepatic biliary atresia. Patients with clinical bleeding may benefit temporarily from replacement of coagulation proteins with FFP, cryoprecipitate, or exchange transfusion. However, without recovery of hepatic function, replacement therapy is futile. VK should be administered to infants in whom cholestatic liver disease is suspected.
von Willebrand Disease
Although von Willebrand disease (VWD) is the most common inherited bleeding disorder, patients with the disease rarely present in the neonatal period. Plasma concentrations of VWF are increased in neonates, as is the proportion of ULVWF multimers. Nonetheless, severe deficiency (type 3) and some qualitative VWF disorders (type 2) have been reported to present in the newborn period. Management of VWD is discussed in detail in Chapter 31 . Use of arginine vasopressin in the newborn period is contraindicated because of associated excess free water retention.
Platelet Disorders
Quantitative Platelet Disorders.
The definition of thrombocytopenia in newborns is the same as that in adults: a platelet count less than 150 × 10 9 /L. Consequently, platelet counts less than 150 × 10 9 /L are abnormal and indicate the need for investigation and, sometimes, treatment. Mean platelet volumes in newborns are similar to those of adults (range, 7 to 9 fL). Postnatally, the mean platelet volume increases slightly over the first 2 weeks of life, concomitantly with an increase in platelet count.
Epidemiology.
Thrombocytopenia is the most common hemostatic abnormality in newborns admitted to neonatal intensive care units (NICUs). A single prospective cohort study and six retrospective reviews have provided the most reliable information on the occurrence, natural history, mechanisms, and clinical impact of thrombocytopenia in newborns. There is general agreement that thrombocytopenia indicates the presence of an underlying pathologic process; however, the clinical relevance of mild thrombocytopenia is unknown. Thrombocytopenia develops in approximately 22% to 35% of infants and approximately 75% of extremely low birth weight neonates admitted to the NICUs of tertiary hospitals. In some infants the thrombocytopenia is trivial, with platelet counts between 100 and 150 × 10 9 /L. However, in 50% of affected infants, platelet counts decrease to less than 100 × 10 9 /L, and in 20% of infants, platelet counts are lower than 50 × 10 9 /L. What constitutes a safe platelet count in a neonate remains to be rigorously determined, but many neonatal units attempt to keep the platelet count higher than 50 × 10 9 /L, although this figure may vary, depending on the postconceptional age of the infant and comorbid conditions.
Pathogenesis.
Causes of thrombocytopenia in neonates, like those in older children and adults, can be divided into disorders involving increased platelet destruction, decreased platelet production, or sequestration ( Box 5-1 ). Although many of these causes overlap with those seen in older children and adults, several entities are unique to neonates or may occur in the newborn period and are considered in more detail here. Increased platelet destruction is the mechanism responsible for thrombocytopenia in most infants. Characterization of mechanisms responsible for thrombocytopenia is important because they have practical implications in assessing the risk for bleeding and in guiding management.
INCREASED DESTRUCTION OF PLATELETS
Immune-mediated destruction
Neonatal alloimmune thrombocytopenia
Maternal idiopathic thrombocytopenic purpura
Drug-dependent antibody (could be from maternal medication use)
Autoimmune thrombocytopenic purpura
Non–immune-mediated destruction
Pseudothrombocytopenia (platelet clumping) in vitro
Disseminated intravascular coagulation
Asphyxia and placental insufficiency
Perinatal aspiration
Necrotizing enterocolitis
Hemangiomas (Kasabach-Merritt syndrome)
Thrombosis (may be occult)
Respiratory distress syndrome
Maternal preeclampsia
Cardiopulmonary bypass (including extracorporeal membrane oxygenation [ECMO])
Familial thrombotic thrombocytopenic purpura
Other syndromes of destruction
Hyperbilirubinemia
Phototherapy
Polycythemia
Rh hemolytic disease
Total parenteral nutrition
DECREASED PRODUCTION OF PLATELETS
Acquired disorders
Viral infection (e.g., human immunodeficiency virus, cytomegalovirus, rubella virus, human herpesvirus 6)
Congenital leukemia
Down syndrome–transient myeloproliferative disorder (DS-TMD)
Neuroblastoma
Histiocytosis
Osteopetrosis
Inherited disorders
Disorders associated with small platelets
Wiskott-Aldrich syndrome (WAS)
X-linked thrombocytopenia (associated with Wiskott-Aldrich syndrome protein [WASP])
Disorders associated with normal-size platelets
Thrombocyopenia with absent radii (TAR)
Amegakaryocytic thrombocytopenia with radioulnar synostosis (ALTRUS)
Congenital amegakaryocytic thrombocytopenia (CAMT)
Familial platelet disorder with propensity for development of acute myelogenous leukemia (FPD/AML)
ANKRD26-related thromobocytopenia (THC2)
Cytochrome C–related thrombocytopenia (CYCS)
Disorders associated with large platelets
X-linked macrothrombocytopenia with or without anemia ( GATA-1 mutations)
MYH9-related disorders (May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, Epstein syndrome)
Bernard-Soulier syndrome
Paris-Trousseau/Jacobsen syndrome
Velocardiofacial syndrome
Montreal platelet syndrome
Gray platelet syndrome
Platelet-type von Willebrand diseaes (pseudo–von Willebrand disease)
Filamin-related thrombocytopenia
Tubulin-related thrombocytopenia
Thrombocytopenia associated with sitosterolemia
Other disorders
Fanconi anemia
Metabolic disorders (e.g., methylmalonic acidemia, ketotic glycinemia, isovaleric acidemia)
Drug-mediated suppression
SEQUESTRATION
Hypersplenism
Increased Platelet Destruction.
Thrombocytopenia secondary to increased platelet destruction can be divided into immune and nonimmune causes.
Immune Thrombocytopenia.
Immune thrombocytopenia is defined as an increased rate of platelet clearance caused by platelet-associated immunoglobulin G (IgG) or complement. It is the most common form of increased platelet destruction in newborns and should always be suspected in otherwise healthy infants with isolated severe thrombocytopenia. Neonatal immune thrombocytopenia can be further broken down into one of three processes: transplacental passage of a maternal antibody directed against a nonshared platelet antigen (termed alloimmune thrombocytopenia or, sometimes, isoimmune thrombocytopenia), transplacental passage of a cross-reactive maternal autoimmune-derived antiplatelet antibody, or generation of an autoreactive antiplatelet antibody by the newborn itself (termed autoimmune thrombocytopenia). The second can be distinguished from the other two by examining the mother for a low platelet count. Differentiation of autoimmune thrombocytopenia from alloimmune thrombocytopenia in neonates is critical because the management and severity of these disorders are quite different. Chapter 34 discusses these two forms of immune thrombocytopenia and their management in detail.
Neonatal Alloimmune Thrombocytopenia.
Neonatal alloimmune thrombocytopenia (NAIT) is the most common cause of immune-mediated thrombocytopenia in the newborn period, with an estimated incidence of about 1 in 1000 to 5000 live births in white populations. It arises when the mother, who lacks a common platelet antigen, is exposed to the antigen (inherited from the father) on neonatal platelets and generates a neutralizing IgG. This crosses the placenta and mediates premature clearance of fetal and newborn platelets. First-born infants can be affected. Frequently, the thrombocytopenia is severe. It is critical to make the diagnosis because the risk of life-threatening bleeding and central nervous system morbidity is high, estimated at up to 10% to 15% of cases. A useful diagnostic test is to assay the reactivity of the mother’s serum with the father’s platelets. If such samples are not available, it is possible to look for reaction of the mother’s serum with the newborn’s platelets or the presence of antiplatelet antibodies in the newborn’s serum. The most common antigen is HPA-1a (formerly called Pl A1 ), although other antigens, such as HPA-3a (formerly called Bak a ), HPA-4a (formerly called Pen a [more common in Asian populations]), HPA-5a (formerly called Br b ), and HPA-5b (formerly called Br a ), have been described. Not all cases in which the pregnant mother generates allosensitive antiplatelet antibodies result in neonatal thrombocytopenia.
Treatment of NAIT includes transfusion with washed platelets donated by the mother. Because the mother’s platelets lack the offending antigen, they should have a normal life span. Many blood banks also maintain registries of HPA-1a–negative donors who can be contacted for platelet donation in cases involving antibodies against HPA-1a. If antigen-negative platelets are not available, treatment with intravenous immunoglobulin, 1 g/kg as a single daily bolus for 1 or 2 consecutive days, along with random donor platelets if the patient is bleeding, is effective in reducing the rate of platelet destruction. Endogenous platelet counts typically rise by 36 to 48 hours. The role of corticosteroids in the treatment of infants with NAIT is unclear, and they are typically not used as initial therapy. All infants with NAIT should undergo head ultrasonography or computed tomography to rule out ICH. The natural history of NAIT is gradual resolution of the thrombocytopenia over the first few months of life as the offending antibody is cleared. Close monitoring of the platelet count is indicated for at least the first month of life until the platelet count normalizes. The safety of breastfeeding in the setting of NAIT has not been rigorously studied. One case report suggested that breastfeeding did not significantly worsen the clinical course, even though the breastmilk contained antiplatelet IgG.
Immune Thrombocytopenia Caused by Maternal Antiplatelet Autoantibodies. Maternal autoimmune antiplatelet IgG can cross the placenta and result in increased platelet destruction in the fetus. A common cause is maternal systemic lupus erythematosus. Like NAIT, the thrombocytopenia gradually resolves over the first few months of life as the maternal antibodies are cleared. In severe cases, use of intravenous immunoglobulin in the neonate may help reduce the rate of platelet destruction. Corticosteroids are of no clear utility. Drug-dependent antibodies can result in both maternal and neonatal thrombocytopenia.
Autoimmune Thrombocytopenia.
True autoimmune thrombocytopenia, in which neonates generate an antibody against their own platelets, is very rare in the newborn period. If present, it typically signals an underlying immune dysregulatory disorder. If heparin-induced thrombocytopenia is suspected, heparin therapy should be discontinued immediately and alternative forms of anticoagulation considered if necessary (see Chapter 34 ).
Nonimmune Thrombocytopenia
Sepsis and Disseminated Intravascular Coagulation.
A common nonimmune cause of thrombocytopenia in the newborn period is sepsis, with or without DIC. Viral infections (rubella virus, herpesvirus, echovirus, cytomegalovirus, human herpesvirus 6, and human immunodeficiency virus), protozoal infections (Toxoplasma), and bacterial infections cause severe thrombocytopenia. Mechanisms responsible for bacterial sepsis–induced thrombocytopenia are multifactorial and include consumption resulting from DIC, endothelial damage, platelet aggregation caused by binding of bacterial products to platelet membranes, and decreased production as a result of marrow infection. Mechanisms responsible for virus-induced thrombocytopenia include loss of sialic acid from platelet membranes because of viral neuraminidase, intravascular platelet aggregation, and impaired megakaryopoiesis (see later). Congenital rubella causes thrombocytopenia in three quarters of infants, with platelet counts ranging from 20 to 60 × 10 9 /L for the first 4 to 8 weeks of life.
In premature infants, thrombocytopenia often complicates other disorders such as RDS, persistent pulmonary hypertension, necrotizing enterocolitis, and hyperbilirubinemia treated by phototherapy. Activation of coagulation with platelet consumption occurs in RDS, and mechanical ventilation may be an independent factor contributing to thrombocytopenia. It has been suggested that persistent pulmonary hypertension in newborns may be due in part to intrapulmonary platelet aggregation and the release of platelet-derived vasoactive substances such as thromboxane A 2 . Approximately half of infants with necrotizing enterocolitis are thrombocytopenic, with about 20% having laboratory evidence of DIC.
Exchange Transfusion, Hyperbilirubinemia, and Phototherapy.
Intrauterine and exchange transfusions cause thrombocytopenia by a dilutional effect that depends on the amount of blood transfused. After exchange transfusion, platelet counts increase within 3 days and reach preexchange levels by about 7 days. Both hyperbilirubinemia and phototherapy are associated with mild thrombocytopenia in newborn humans and shortened platelet survival in rabbits.
Hypoxia and Placental Insufficiency.
Acute asphyxia is a consistent cause of DIC and thrombocytopenia. Chronic hypoxia is associated with placental dysfunction, intrauterine growth retardation, and significant thrombocytopenia.
Vascular Malformations.
Certain types of large vascular malformations are associated with Kasabach-Merritt syndrome, in which a local consumptive coagulopathy occurs as a result of abnormal endothelium. This causes hypofibrinogenemia, elevated levels of fibrinogen-fibrin degradation products, microangiopathic fragmentation of red cells, and thrombocytopenia (see Chapter 34 ). The thrombocytopenia can be severe, with platelet counts lower than 50 × 10 9 /L. Approximately 50% of affected infants experience systemic bleeding during the first month of life. Sometimes the vascular malformations are not apparent on physical examination and require imaging studies to detect.
Thrombosis and Familial Thrombotic Thrombocytopenic Purpura.
Thrombocytopenia is loosely associated with thromboembolic complications and polycythemia in infants with hematocrit values greater than 70%. However, thrombocytopenia may also indicate the presence of other concurrent disease processes. Infants with a familial form of hemolytic-uremic syndrome or thrombotic thrombocytopenic purpura have a microangiopathic hemolytic anemia in association with transient neurologic or renal abnormalities (see Chapter 34 ).
Decreased Platelet Production.
Thrombocytopenia secondary to decreased platelet production is rare and accounts for less than 5% of thrombocytopenic infants. Causes include viral infections, drug-induced thrombocytopenia, congenital leukemia, Down syndrome transient myeloproliferative disorder (DS-TMD), neuroblastoma, histiocytosis, osteopetrosis, congenital amegakaryocytic thrombocytopenia (CAMT), thrombocytopenia with absent radius syndrome (TAR), other inherited genetic platelet disorders, and bone marrow failure syndromes.
Congenital Viral Infection.
Congenital viral infections with rubella virus, herpesvirus, echovirus, cytomegalovirus, human herpesvirus 6, and human immunodeficiency virus have been associated with thrombocytopenia in the newborn period, in part through deleterious effects on megakaryopoiesis and thrombopoiesis.
Drug-Induced Thrombocytopenia.
Transplacental passage of drugs or the use of drugs in neonates can cause thrombocytopenia via bone marrow suppression or the development of drug-dependent antibodies, or both. However, these causes are rare, and evidence for them is weak. Agents implicated are salicylates, quinine, hydralazine, tolbutamide, and thiazide diuretics.
Congenital Leukemia and Other Malignant Diseases.
Infantile leukemia, as well as other neonatal malignant diseases characterized by bone marrow infiltration, such as neuroblastoma, can be accompanied by thrombocytopenia in the newborn period. Typically, other hematopoietic lineages are affected and other clinical signs are apparent. An infantile form of myelofibrosis has been described in several families in Saudi Arabia.
Down Syndrome–Transient Myeloproliferative Disorder.
Some infants with Down syndrome (DS) (trisomy 21) are born with a transient myeloproliferative disorder (DS-TMD) characterized by leukocytosis, thrombocytopenia, predominance of circulating early erythromegakaryocytic precursor cells, pancytopenia, and, in some cases, severe liver fibrosis. Remarkably, this myeloproliferation resolves spontaneously over the first few months of life. In about 20 % of symptomatic cases, acute megakaryocytic leukemia (DS-AMKL) develops within a few years. The DS-TMD and DS-AMKL cells harbor acquired mutations in the gene encoding the erythromegakaryocytic transcription factor GATA-1.
Inherited Platelet Disorders.
Though considerably less common than consumptive processes or viral suppression, inherited platelet disorders can occur in the newborn period and should be considered in the differential diagnosis of thrombocytopenia in a neonate. They are discussed in more detail in Chapter 30 (also see Box 5-1 for a complete listing). Several classifications, diagnostic algorithms, and management recommendations have been published. Infants with aplastic disorders have the greatest risk of serious bleeding in the form of ICH in the first months of life. Brief descriptions of selected syndromic disorders with specific neonatal manifestations are given here.
Disorders with Small Platelets
Wiskott-Aldrich Syndrome.
Wiskott-Aldrich syndrome (WAS; OMIM 301000) is an X-linked disorder caused by deficiency of the Wiskott-Aldrich syndrome protein, which functions in actin dynamics in megakaryocytes and T lymphocytes. It is one of the few disorders characterized by small platelets. The small size of the platelets is not always apparent by standard automated measurements of blood cell mean platelet volume. Therefore if the diagnosis is being considered, it is imperative to evaluate platelet size by examination of the peripheral smear and to confirm small platelet volume with cytometer devices calibrated for platelets. The other clinical hallmarks of WAS are immunodeficiency (usually severe) and eczema. Bloody diarrhea is often seen and can be an initial sign. The immunodeficiency is frequently the most significant clinical problem associated with this disease. It is important to recognize the syndrome early so that appropriate interventions can be instituted, including prophylactic treatment against opportunistic infections such as Pneumocystis carinii infection. A destructive component of the thrombocytopenia in WAS is also likely because splenectomy increases platelet counts in a large number of patients. In addition, the thrombocytopenia can be exacerbated by development of immune thrombocytopenic purpura secondary to the immune dysregulation. Bone marrow transplantation is curative for WAS, and early diagnosis is helpful in initiating listing for transplantation before significant complications arise.
X-Linked Thrombocytopenia.
X-linked thrombocytopenia (OMIM 313900) is a less severe form of WAS that is manifested predominantly as thrombocytopenia. Typically, there is no or minimal immunologic dysfunction. However, more severe immunologic disturbances may develop in some patients over time. This clinical entity is distinct from X-linked macrothrombocytopenia, which is associated with germline mutations in GATA-1.
Disorders with Normal-Size Platelets
Congenital Amegakaryocytic Thrombocytopenia.
Congenital amegakaryocytic thrombocytopenia (CAMT; OMIM 604498) is characterized by severe thrombocytopenia with a markedly reduced number or absence of megakaryocytes. Patients are typically seen shortly after birth with petechiae, bruising, or bleeding. Biallelic inactivating mutations in the gene for c-mpl, the receptor for the major megakaryocyte cytokine TPO, have been identified in a large number of patients. Interestingly, no patients have been reported to date with mutations in the TPO gene itself. Patients with severe CAMT are at high risk for the development of complete bone marrow failure, usually within the first few years of life. Such failure is most likely due to a role of TPO signaling in hematopoietic stem cell function. Bone marrow transplantation has been used successfully to treat patients with severe CAMT.
Thrombocytopenia with Absent Radii.
Thrombocytopenia with absent radii syndrome (TAR; OMIM 274000) is characterized by hypomegakaryocytic thrombocytopenia and bilateral radial hypoplasia with the presence of thumbs. Other hematopoietic lineages are not affected. Additional skeletal features may include shortening or, rarely, aplasia of the humerus and ulna. The lower limbs are frequently affected but to a lesser extent. Congenital cardiac disease and cow’s milk intolerance have also been associated in some cases. The genetic basis of TAR has recently been clarified. Common interstitial microdeletions of 200 kb on chromosome 1q21.1 have been detected in several families with TAR, and the key gene within this region is thought to be RBM8A, which encodes a subunit of the exon junction complex involved in RNA processing. Microdeletion or monoallelic null mutations in RBM8A are typically insufficient to cause disease. However, coinheritance of a low-frequency single-nucleotide polymorphism in an RBM8A gene regulatory sequence on the opposite allele is associated with disease penetrance.
Neither splenectomy nor steroids are beneficial for infants with TAR. Platelet transfusions are highly effective but should be reserved for symptomatic infants because prophylactic platelet transfusions could result in refractoriness as a result of allosensitization. By several months of age, increased numbers of megakaryocytes usually appear in the bone marrow, and platelet counts increase. A functional platelet defect may be present in some children with TAR.
Amegakaryocytic Thrombocytopenia with Radioulnar Synostosis.
Amegakaryocytic thrombocytopenia with radioulnar synostosis (ATRUS; OMIM 605432) is a rare disorder characterized by reduced or absent megakaryocytes, severe thrombocytopenia, possible aplastic anemia, and proximal radioulnar synostosis leading to difficulty with forearm pronation. Sensorineural hearing loss may also be present. Mutations in the gene encoding the homeobox containing transcription factor HOXA11 have been found in some familial cases of ATRUS.
Disorders with Large Platelets
Paris-Trousseau Syndrome or Jacobsen Syndrome.
Paris-Trousseau syndrome (OMIM 188925) (also called Jacobsen syndrome; OMIM 147791) is a constitutional contiguous gene deletion disorder involving the long arm of chromosome 11 (11q23). The constellation of findings in this syndrome includes severe congenital cardiac abnormalities, trigonocephaly, mental retardation, dsymorphogenesis of the hands and face, and macrothrombocytopenia. The cause of the thrombocytopenia appears to be related to impaired platelet production because platelet survival time is normal. These patients can also exhibit a predisposition to thrombosis, thus suggesting possible qualitative defects in platelet production. Examination of bone marrow reveals significant dysmegakaryopoiesis with an abundance of micromegakaryocytes and death of large numbers of megakaryocytes during the late stages of maturation. Peripheral blood platelets contain giant alpha granules, which are thought to arise from aberrant alpha-granule fusion during prolonged residence in bone marrow. The minimal chromosome regions deleted in Paris-Trousseau or Jacobsen syndrome associated with thrombocytopenia include genes for the Ets family transcription factors FLI-1 and ETS-1, both of which play roles in megakaryocyte development. Lentiviral expression of FLI-1 in CD34+ cells from patients with Paris-Trousseau thrombocytopenia rescues megakaryocyte differentiation in vitro, thereby providing evidence that FLI-1 deficiency is the cause of impaired thrombopoiesis in these patients.
Velocardiofacial Syndrome.
Velocardiofacial syndrome (OMIM 192430) is a contiguous gene deletion disorder involving chromosome 22q11.2. Clinical manifestations include cleft palate, cardiac anomalies, typical facies, learning disabilities, and defective GPIb/IX/V function. The platelet effects are due to heterozygous loss of the GPIbβ gene, which resides in this region, and the condition should be considered a mild form of Bernard-Soulier disorder.
Hypersplenism.
As in adults, thrombocytopenia resulting from hypersplenism is usually mild to moderate, with platelet counts ranging from 50 to 100 × 10 9 /L in newborns.
Clinical Impact of Neonatal Thrombocytopenia.
Clinically important bleeding is less likely to occur in patients with consumptive platelet disorders than in those with production defects. The bleeding risk is increased in patients who have both thrombocytopenia and a defect in platelet function. Selection of a platelet count at which one should intervene, though a simplistic response, provides a guideline for therapy. A platelet count less than 50 × 10 9 /L places some otherwise healthy full-term newborns at risk for serious ICH. The importance of “moderate” thrombocytopenia (platelet counts between 50 and 100 × 10 9 /L) in sick premature infants has been a subject of controversy. A randomized, controlled trial assessed the potential benefits of platelet concentrate transfusions in 154 premature thrombocytopenic infants during the first 72 hours of life. Treated infants received platelet concentrates to maintain platelet counts higher than 150 × 10 9 /L. No beneficial effect on the incidence or extension of ICH was shown in this study, which was designed to detect an effect of 25% or greater. However, infants who received transfusions had shortened bleeding times and required significantly less blood product support. A retrospective study of 53 neonates with severe thrombocytopenia treated at a single NICU showed no significant difference in major hemorrhage in those transfused to maintain a platelet count greater than 30 × 10 9 /L versus greater than 50 × 10 9 /L.
Treatment.
Management of thrombocytopenic infants depends in part on the underlying disorder. If an infant is bleeding, a trial of platelet concentrates (10 to 20 mL/kg) is indicated. The increased platelet count usually shortens the bleeding time and is frequently clinically effective. Autoimmune and alloimmune thrombocytopenia typically does not respond to random donor platelet concentrates and requires additional specific forms of therapy.
Thrombocytosis in the Newborn.
Elevated platelet counts are frequently observed in premature infants at approximately 4 to 6 weeks after birth. There are no clinical manifestations of neonatal thrombocytosis, and therapeutic intervention is not indicated.
Qualitative Platelet Disorders.
Despite the physiologic hyporeactivity of neonatal platelets in response to exposure to some agents, healthy infants do not have an increased risk of bleeding. Pathologic impairment of platelet function may occur, to a variable extent, as a result of the use of certain drugs or the presence of pathologic states in either mothers or infants. In mothers, causative factors include the use of some drugs, diabetes, dietary abnormalities, smoking, and ethanol abuse ; in infants, they include the use of some drugs, perinatal aspiration syndrome, hyperbilirubinemia, phototherapy, renal failure, and hepatic failure.
Phototherapy.
When platelets are exposed to a broad-spectrum blue fluorescent light in vitro, aggregation is decreased and microscopic alterations in granules and external membranes occur.
Medications
Aspirin.
Salicylate crosses the placenta and can be detected in fetuses after maternal ingestion. Clearance of salicylate is slower in newborns than in adults, and thus infants have a potential risk of bleeding for a prolonged time. However, in vitro studies have not demonstrated increased sensitivity of newborn platelets to salicylates as compared with adult platelets, and the evidence linking maternal aspirin ingestion to clinically important bleeding in newborns is weak. There is little reason to have serious concerns about maternal ingestion of aspirin, but it is reasonable to advise mothers to not ingest aspirin unless specifically instructed by their physician.
Indomethacin.
Indomethacin is used for nonsurgical closure of a patent ductus arteriosus in premature infants, but it also has antiplatelet activities. Indomethacin, like salicylate, has a longer half-life in newborns than in adults (21 to 24 hours in newborns vs. 2 to 3 hours in adults). This extended half-life is probably due to underdevelopment of hepatic drug metabolism or renal excretory function or to altered protein binding. Indomethacin inhibits platelet function in newborns, as shown by prolongation of bleeding times. Randomized, controlled trials have provided conflicting conclusions on the effect of indomethacin on intraventricular hemorrhage in premature infants.
Maternal Diabetes.
The reactivity of platelets from diabetic mothers and their infants is increased, as demonstrated by enhanced thromboxane B 2 production, enhanced platelet aggregation, and a lower threshold to many aggregating agents. The enhanced platelet function in diabetes is associated with increased synthesis of a prostaglandin E–like substance that crosses the placenta and affects the fetus. Despite these in vitro findings, the evidence linking enhanced platelet reactivity to clinically significant thromboembolism in newborns of diabetic mothers is weak.
Diet.
Alterations in the diet of mothers or infants during the postnatal period can affect newborn platelet function. Increases in the ratio of polyunsaturated fatty acids to saturated fatty acids in the diet of mothers who breastfeed their infants result in increases in the concentration of linoleic acid and enhanced thromboxane B 2 production. Infants receiving a diet deficient in essential fatty acids may exhibit arachidonic acid depletion and platelet dysfunction. Vitamin E functions as an antioxidant and as an inhibitor of platelet aggregation/release in humans. Instances of vitamin E–deficient infants with increased platelet aggregation that reversed after vitamin E supplementation have been reported.
Amniotic Fluid.
Amniotic fluid contains procoagulant activity that enhances the generation of thromboxane A 2 by platelets. Infants in whom perinatal aspiration syndrome develops have pulmonary hypertension with platelet thrombi in the pulmonary microcirculation. The exact mechanism or mechanisms leading to persistent pulmonary hypertension in these infants is unknown.
Nitric Oxide.
Nitric oxide prevents adhesion of platelets to endothelial cells and inhibits ADP-induced aggregation of cord platelets in a manner similar to that in adults.
Extracorporeal Membrane Oxygenation.
Extracorporeal membrane oxygenation (ECMO) permits transfer of oxygen into blood across a semipermeable membrane and is currently used for infants with life-threatening severe respiratory insufficiency. Underlying respiratory disorders include meconium aspiration syndrome, severe RDS, congenital diaphragmatic hernia, persistent pulmonary hypertension, and sepsis. Hemorrhage, particularly ICH, is one of the most serious complications of this technique. Hardart and Fackler reported an overall incidence of new ICH of about 10% in infants included in the Extracorporeal Life Support Organization Registry (N = 4550) from 1992 through 1995. Of 1398 evaluable premature infants (born at <37 weeks’ EGA) reported to the registry between 1992 and 2000, ICH developed in 13%. Similar to that found with cardiopulmonary bypass, the increased risk of bleeding during ECMO is due mostly to the use of heparin in combination with other hemostatic defects, including significantly decreased plasma concentrations of coagulation factors and platelet dysfunction secondary to chronic activation. Other recognized contributing factors include prolonged hypoxia, ischemia, general anesthesia, acidosis, sepsis, and treatment with epinephrine. In premature infants, the risk for ICH with ECMO correlates inversely with postconceptional age. Although anticoagulation is required for ECMO, the optimal use of heparin has never been tested in clinical trials.
Thrombotic Disorders in Neonates
Congenital Prothrombotic Disorders
Patients with heterozygous defects for recognized inherited prothrombotic disorders are rarely seen with their first thromboembolic complication during childhood unless another pathologic event unmasks the problem. In contrast, patients who are homozygotes or double heterozygotes for a congenital prothrombotic disorder are often initially encountered in the newborn period or during early childhood. The following discussion is limited to the unique aspects of these inherited deficiencies in newborns. Chapter 33 discusses congenital prothrombotic disorders in detail.
Homozygous Prothrombotic Disorders.
Though rare, the most commonly reported homozygous prothrombotic disorder encountered during the newborn period is protein C deficiency. Homozygous protein S deficiency is even less common. All patients seen in the newborn period had undetectable levels of protein C (or protein S), whereas children with a delayed manifestation had detectable levels ranging between 0.05 and 0.20 U/mL. Rare cases of homozygous AT deficiency have been reported in newborns.
Clinical Findings.
The classic clinical manifestation of homozygous protein C/protein S deficiency consists of cerebral or ophthalmic damage (or both) in utero, purpura fulminans within hours or days of birth, and, on rare occasion, large-vessel thrombosis. Purpura fulminans is an acute, lethal syndrome of DIC characterized by rapidly progressive hemorrhagic necrosis of the skin secondary to dermal vascular thrombosis. The skin lesions start as small, ecchymotic sites that increase in radial fashion, become purplish black with bullae, and then turn necrotic and gangrenous. The lesions develop mainly on the extremities but can occur on the buttocks, abdomen, scrotum, and scalp. They also occur at pressure points, at sites of previous puncture, and at previously affected sites. Moreover, affected infants have severe DIC with hemorrhagic complications. Large-vessel arterial thromboses have been described in a neonate with homozygous AT deficiency.
Diagnosis.
The diagnosis of homozygous protein C/protein S deficiency in infants is based on the appropriate clinical picture, a protein C/protein S level that is usually undetectable, a heterozygous state in the parents, and, ideally, identification of the molecular defect. The presence of very low levels of protein C/protein S in the absence of clinical manifestations and a family history cannot be considered diagnostic, because physiologic plasma levels can be as low as 0.12 U/mL. Molecular diagnosis is available for identified families (see Chapter 33 ).
Initial Treatment.
The diagnosis of homozygous protein C/protein S deficiency is usually unanticipated and made at the time of clinical evaluation. Although numerous forms of initial therapy have been used, 10 to 20 mL/kg of FFP every 6 to 12 hours is usually the form of therapy that is most readily available. Plasma levels of protein C achieved with these doses of FFP vary from 15% to 32% at 30 minutes after the infusion and from 4% to 10% at 12 hours. Plasma levels of protein S (which was entirely bound to C4b) were 23% at 2 hours and 14% at 24 hours, with an approximate half-life of 36 hours.
A protein C concentrate (Ceprotin) received approval by the Food and Drug Administration in 2007 for use in treating congenital protein C deficiency. A second plasma-derived concentrate (Protexel) is also available in Europe. Recommended initial doses for acute thrombotic episodes and short-term prophylaxis in patients with severe protein C deficiency are 100 to 120 IU/kg in neonates, with subsequent doses of 60 to 80 IU/kg every 6 hours and maintenance doses of 45 to 60 IU/kg thereafter every 6 to 12 hours. However, dosing should be adjusted to maintain trough protein C activity of 50%. After resolution of the acute event, the patient should continue on a dose to maintain the trough protein C activity level above 25% for the duration of treatment. Replacement therapy should be continued until all the clinical lesions resolve, which usually takes place at 6 to 8 weeks. In addition to the clinical course, plasma D-dimer concentrations may be useful for monitoring the effectiveness of protein C replacement.
Long-Term Therapy.
Modalities used for the long-term management of infants with homozygous protein C/protein S deficiency include oral anticoagulation therapy, replacement therapy with either FFP or protein C concentrate, or liver transplantation. When oral anticoagulation therapy is initiated, replacement therapy should be continued until the international normalized ratio (INR) is at a therapeutic value so that skin necrosis can be avoided. The therapeutic range for the INR can be individualized to some extent but is usually between 2.5 and 3.5. Risks associated with oral anticoagulation therapy include bleeding with high INR values and recurrent purpuric lesions with low INR values. Frequent monitoring of INR values is required if these complications are to be avoided. Bone development should also be monitored because the long-term effect of warfarin use on the bones of young infants is unknown. Long-term use of low–molecular-weight heparin (LMWH) could be considered, but it also has the potential for osteopenia. Moreover, it is expensive and requires twice-daily subcutaneous injections.
Heterozygous Prothrombotic Disorders.
Thromboembolic events related to heterozygous genetic prothrombotic disorders rarely occur in infants. When they do, a secondary, acquired insult is usually present. The few case reports in the literature describe a diversity of clinical manifestations that usually reflect the site of the thrombus. Purpura fulminans did not occur in any case.
Treatment.
Treatment consists of supportive therapy alone, anticoagulation with heparin, thrombolytic therapy, and replacement with specific factor concentrates. Removal or treatment of the secondary acquired insult is important. For AT deficiency, AT concentrates were administered to four infants as either boluses or continuous infusions. Boluses of 52 and 104 U/kg of AT concentrate increased AT levels from 0.10 U/mL to 0.75 and 1.48 U/mL, respectively, at 1 hour. At 24 hours, levels of AT had decreased to approximately 0.20 U/mL. A continuous infusion of AT concentrate at a rate of 2.1 U/kg/hr maintained a plasma level of 0.40 to 0.50 U/mL.
Acquired Prothrombotic Disorders
Symptomatic secondary thromboembolic complications occur more frequently in sick newborns than in children of any other age, with an incidence of approximately 2.4 per 1000 hospital admissions to the NICU. Intravascular catheters are responsible for more than 80% of venous and 90% of arterial thrombotic complications. Their contribution to thrombosis is probably multifactorial, including the presence of a foreign surface, endothelial cell damage, impairment of flow, and infusion of noxious substances. Renal vein thrombosis (RVT) is the most common form of non–catheter-related thrombosis. Other risk factors include increased blood viscosity because of a high hematocrit, poor deformability of physiologically large red cells, dehydration, and activation of the coagulation and fibrinolytic systems secondary to a variety of medical problems. Very low AT levels have been observed in preterm infants with RDS, which probably contributed to their increased risk for thromboembolic complications.
Venous Catheter–Related Thrombosis.
The use of umbilical venous catheters and other forms of central venous catheters is associated with a significant risk for thrombosis. According to autopsy studies, 20% to 65% of infants who die with an umbilical venous catheter in place have an associated thrombus. Appropriate placement of umbilical venous catheters is critical to prevention of serious organ impairment, such as portal vein thrombosis and hepatic necrosis. Long-term sequelae of umbilical venous catheters have not been rigorously studied but include portal vein thrombosis with portal hypertension, splenomegaly, gastric and esophageal varices, and hypertension. Until recently, pulmonary embolism was rarely diagnosed in sick newborns because its clinical signs were easily confused with those of RDS. Ventilation lung scintigraphy and computed tomography pulmonary angiography have been used in newborns to facilitate the diagnosis of pulmonary embolism, but there are no studies to date that have determined their sensitivity and specificity in this setting.
Arterial Catheter–Related Thrombosis.
Seriously ill infants require indwelling arterial catheters, which are associated with a risk of thrombosis regardless of the vessel or catheter type chosen. Catheter-related thrombosis not only occludes catheters but may also obstruct major arterial vessels. In a retrospective examination of approximately 4000 infants who underwent umbilical artery catheterization, 1% had severe symptomatic vessel obstruction. Asymptomatic catheter-related thrombi occur more frequently, as evidenced by postmortem (3% to 59% of cases) and angiographic (10% to 90% of cases) studies.
Diagnosis.
Contrast-enhanced angiography is considered the reference test for the diagnosis of arterial thrombosis. Noninvasive techniques such as Doppler ultrasound offer advantages, but their sensitivity and specificity are unknown. A review of 20 neonates with aortic thrombosis treated in one institution revealed that ultrasound failed to identify thrombi in four patients, three of whom had complete aortic obstruction.
Sequelae.
The sequelae of catheter-related thrombosis can be immediate or long term. Acute symptoms reflect the location of the catheter and include renal hypertension, intestinal necrosis, and peripheral gangrene. The long-term side effects of symptomatic and asymptomatic thrombosis of major vessels have not been studied but are probably significant.
Prophylaxis with Heparin.
A low-dose continuous heparin infusion (3 to 5 U/hr) is commonly used to maintain catheter patency. The effectiveness of heparin was assessed in seven studies focusing on three outcomes: patency, local thrombus, and ICH. Patency, which is probably linked to the presence of local thrombus, is prolonged with the use of low-dose heparin. Local thrombosis was assessed by ultrasound in two randomized studies. The evidence linking low-dose heparin prophylaxis to ICH in newborns is weak. One study had a sample size of only 15 per arm ; another case-control study had a broad odds ratio that ranged from 1.4 to 11 ; and a third study was a retrospective association analysis that may be confounded by other variables. A randomized study involving 113 premature infants using a 1-U/hr continuous heparin infusion showed no difference in ICH. Thus the magnitude of risk for ICH is uncertain. Heparin is used in at least three quarters of American nurseries.
Renal Vein Thrombosis.
RVT occurs primarily in newborns and young infants. Approximately 80% of cases occur within the first month and usually within the first week of life. In some infants RVT develops in utero. The incidence in male and female infants is similar, and the left and right sides are affected equally. Bilateral thrombosis occurs in 24% of pediatric RVT patients.
Clinical Findings and Etiology.
The initial symptoms and clinical findings are different in neonates and older patients and are influenced by the extent and rapidity of thrombus formation. Neonates generally have a flank mass, hematuria, proteinuria, thrombocytopenia, and impaired function of the involved kidney. Clinical findings suggesting acute inferior vena cava thrombosis include cold, cyanotic, and edematous lower extremities. RVT results from pathologic states characterized by reduced renal blood flow, increased blood viscosity, hyperosmolality, or hypercoagulability.
Coagulation Abnormalities.
The most common coagulation abnormality in RVT is thrombocytopenia, which is usually mild, with average values of 100 × 10 9 /L. Thus RVT should be considered in the differential diagnosis of thrombocytopenia in neonates. Coagulation may be prolonged and levels of fibrinogen-fibrin degradation products increased. Children with RVT are often evaluated for a congenital prothrombotic disorder, although the significance of thrombophilic markers in this disease is unknown.
Diagnosis and Treatment.
Ultrasound is the radiographic test of choice for the diagnosis of RVT because of ease of testing and sensitivity to an enlarged kidney. Treatment options include supportive care, anticoagulation, and thrombolytic therapy. The use of anticoagulants and thrombolytic agents is controversial. Evidence-based guidelines have recently been published. Either supportive care with serial radiographic monitoring or heparin therapy with therapeutic doses for 6 weeks to 3 months should be considered for unilateral RVT in the absence of uremia and extension into the inferior vena cava. Heparin therapy with therapeutic doses for 6 weeks to 3 months is recommended for unilateral RVT that extends into the inferior vena cava or for bilateral RVT because of the risk of pulmonary embolism and complete renal failure. Thrombolytic therapy should be considered in the presence of bilateral RVT and renal failure. Thrombectomy, though a common therapeutic choice in the past, is rarely indicated.
Outcome.
RVT has changed from a frequently lethal complication to one that is now associated with nearly 100% survival. However, there are considerable long-term renal sequelae, with approximately 70% of affected kidneys having irreversible atrophy, approximately 20% of children having hypertension, and approximately 3% of children having chronic renal failure.
Spontaneous Venous and Arterial Thrombosis.
Spontaneous venous thrombosis occurs in the adrenal veins, inferior vena cava, portal vein, hepatic veins, and venous system of the brain. Spontaneous occlusion of arterial vessels in the absence of a catheter is unusual, but it can occur in ill infants. As in catheter-related thrombosis, the clinical findings reflect the vessel that is occluded. Complete occlusion of a vessel can lead to gangrene and loss of the affected limb or to ischemic organ damage. The presence of systemic hypertension in newborns is frequently related to renal artery thrombosis, even in the absence of a catheter.
Anticoagulation Therapy in Newborns
Heparin Therapy in Newborns.
Guidelines for antithrombotic therapy in neonates have been published. However, the limited strength of the recommendations reflects the lack of controlled trials in this area. Recommendations for adult patients provide useful guidelines but probably do not reflect the optimal therapy for newborns. Current therapeutic options include supportive care alone, anticoagulant therapy, thrombolytic therapy, and thrombectomy. For most infants in whom thrombotic complications develop, the cause of thrombosis is a catheter-related thrombus that does not produce clinical symptoms. In most nurseries, catheters are not routinely checked for associated thrombosis; thus, by exclusion, most infants with clinically silent thrombi receive supportive care alone.
Age-Dependent Features.
Although the benefits of heparin therapy in newborns are probably similar to those in adults, the relative risk that major bleeding will occur with its use may be increased. Infants with thromboses that are extending or infants whose organ or limb viability is threatened by thrombosis may benefit from heparin therapy.
Heparin’s anticoagulant activities are mediated primarily through acceleration of inhibition of thrombin and factor Xa by AT. Although dosing for heparin therapy in newborns differs from that in adults, optimal dosing cannot be predicted. Several observations suggest that the heparin requirements of neonates are decreased in comparison to those of adults. First, the capacity of plasma from healthy newborns to generate thrombin is both delayed and decreased in comparison to that of adult plasma, but similar to that of plasma from adults receiving therapeutic amounts of heparin. Second, at heparin concentrations in the therapeutic range, the capacity of plasma from healthy newborns to generate thrombin is barely measurable. Third, the amount of clot-bound thrombin is decreased in newborns because low plasma concentrations of prothrombin probably reduce heparin requirements. Observations suggesting higher heparin requirements include the following: (1) clearance of heparin is accelerated in newborns, and (2) plasma concentrations of AT are decreased to levels frequently less than 0.40 in premature infants, which may limit heparin’s antithrombotic activities.
Therapeutic Range and Dose.
Therapeutic ranges reflect the optimal risk-benefit ratio of anticoagulant therapy with regard to recurrent thrombotic events and bleeding complications. In the absence of clinical trials in newborns, one approach is to use heparin in doses that achieve the lower therapeutic range for adults (see Chapter 33 ).
General guidelines for initial dosing and subsequent dose adjustment of unfractionated heparin in full-term and premature neonates have been published. Average doses of heparin required in newborns are bolus doses of 25 to 100 U/kg, depending on gestational age, and average maintenance doses of 15 to 28 U/kg/hr, again depending on gestational age. Adjustments are made most reliably with heparin assays, with a target-level anti-Xa range of 0.35 to 0.70 U/mL. Because of the short half-life of unfractionated heparin, simple discontinuation of the infusion is typically sufficient to reverse anticoagulation in the event of hemorrhage. For more rapid reversal, protamine sulfate can be used to neutralize heparin in vivo. However, anaphylactoid reactions with hypotension and bradycardia have been associated with protamine use in children and adults, and protamine should therefore be used with caution.
The use of LMWH offers significant therapeutic advantages over unfractionated heparin, and there is now considerable experience using these preparations in newborns. Potential advantages include more predictable pharmacokinetics, the need for less frequent monitoring than with unfractionated heparin, ease of administration, decreased bleeding, and possibly a lower incidence of heparin-induced thrombocytopenia. Full-term and moderately preterm (28 to 36 weeks EGA) infants metabolize heparin at considerably faster rates than older children and adults do and typically require higher doses to maintain the same levels. Very premature infants (<28 weeks) have similar pharmacokinetic parameters for heparin as do older children and adults. General guidelines for initial therapeutic dosing and subsequent sliding-scale dose adjustments of LMWH in neonates of different gestational ages have been published. Typically, higher doses of LMWH are required in neonates compared with adults to achieve the same anti-Xa levels. Traditional doses of enoxaparin in full-term neonates for full anticoagulation are 1.5 mg/kg given subcutaneously every 12 hours, but they can be as high as 2 mg/kg given subcutaneously every 12 hours in preterm neonates. Levels should be monitored with anti-Xa heparin assays on samples drawn via fresh venipuncture 4 to 6 hours after the subcutaneous dose is given to achieve levels of 0.5 to 1 U/mL for full therapeutic dosing. Levels should be checked every two to four doses for the first week of therapy, then once a week for a month, and then monthly if the infant is stable. Infants who do not respond appropriately to heparin therapy, despite adequate levels, may benefit from infusions of AT concentrate, even if the levels are close to the normal physiologic range for their age. Intramuscular and arterial puncture and the use of antiplatelet medications should be avoided in newborns receiving heparin therapy, and platelet counts should be monitored periodically.
Like unfractionated heparin, LMWH can be reversed with the use of protamine, with dosing dependent on the last dose of LMWH given. As discussed earlier, protamine must be given slowly, and caution must be exercised to avoid potential hypersensitivity reactions. Repeat doses of protamine may be required to reverse the effects of LMWH because the half-life of protamine is relatively shorter than that of LMWH.
Close monitoring of the thrombus with objective means such as ultrasound is recommended. The duration of heparin therapy required for the treatment of thromboembolic complications is uncertain. One approach is to treat the infant for 10 to 14 days with heparin alone. If there is subsequent extension of the thrombus in the absence of anticoagulation therapy, treatment with oral anticoagulants may be considered.
Adverse Effects.
There are two clinically important adverse effects of heparin therapy: hemorrhage, including ICH, and heparin-induced thrombocytopenia. In the absence of an alternative cause, thrombocytopenic patients should be evaluated for heparin-induced thrombocytopenia and treated with alternative therapies.
Oral Anticoagulant Therapy in Newborns
Age-Dependent Features.
Oral anticoagulation therapy in children is discussed in Chapter 33 , and only specific issues related to newborns are discussed in this section. The oral anticoagulant warfarin works by reducing functional plasma levels of the VK-dependent proteins. At birth, levels of the VK-dependent proteins are similar to those found in adults receiving therapeutic amounts of warfarin for deep venous thrombosis/pulmonary embolism. In addition, stores of VK are low, and a small number of newborns have evidence of functional VK deficiency. These features significantly increase the sensitivity of newborns to warfarin and potentially their risk for bleeding. Oral anticoagulant therapy should be avoided when possible during the first month of life. Unfortunately, a small number of infants require extended anticoagulation therapy, and heparin cannot be used for extended periods because of the risk of osteopenia.
Indications, Therapeutic Range, and Dose.
The optimal therapeutic INR range is unknown for newborns and almost certainly differs from that for adults. Recommendations for oral anticoagulation therapy in adults can be used as a guideline for determining the lowest effective dose, which to some extent can be individualized. Maintenance doses for warfarin are age dependent, with infants requiring the highest doses (0.32 mg/kg).
Adverse Effects.
Close monitoring of oral anticoagulation in newborns is required if both hemorrhagic and recurrent thrombotic complications are to be prevented. Unfortunately, these infants often have poor venous access, as well as complicated medical problems. Weekly or biweekly INR measurements and frequent dose adjustments are required. Doses are affected by diet, medications, and intercurrent illnesses. Breastfed infants are very sensitive to oral anticoagulants because of low concentrations of VK in breastmilk. Daily supplementation of breastfed infants with small amounts of commercial formulas reduces their sensitivity to oral anticoagulants and the risk of sudden increases in INR values. In contrast to breastfed infants, infants receiving commercial formulas or total parenteral nutrition are resistant to oral anticoagulants because of VK supplementation. Reducing or removing VK supplementation in infants receiving total parenteral nutrition significantly decreases the dose requirements. Most infants requiring oral anticoagulants also require other medications on an intermittent and long-term basis. The effects of dosage changes and the introduction of new medications must be closely supervised.
Antiplatelet Agents in Newborns.
Antiplatelet agents are rarely used in newborns for antithrombotic therapy. The hyporeactivity of neonatal platelets and the paradoxically short bleeding time suggest that optimal use of antiplatelet agents differs in newborns and adults. Aspirin is the most commonly used antiplatelet agent. The empiric use of low doses of 1 to 5 mg/kg/day has been proposed as adjuvant therapy for patients with Blalock-Taussig shunts, some endovascular stents, and some thrombotic cerebrovascular events. Use of the antiplatelet agent clopidogrel (Plavix) in infants as young as 6 weeks of age has been reported in a small retrospective series. A prospective, multicenter, placebo-controlled pharmacodynamic trial of clopidogrel in children (from birth to 24 months) with a cardiac condition demonstrated that a dose of 0.20 mg/kg/day achieved a platelet inhibition level similar to that in adults receiving a standard dose of 75 mg/day, although there was significant interpatient variability in platelet inhibition levels. Clopidogrel was well tolerated by the infants.
Thrombolytic Therapy in Newborns
Age-Dependent Features.
The activity of thrombolytic agents depends on endogenous concentrations of plasminogen, which are physiologically decreased at birth. Low plasminogen levels result in impairment of the capacity to generate plasmin and a decrease in the capacity to thrombolyse fibrin clots. In addition, fetal glycoforms of plasminogen are less efficiently converted to plasmin by tPA than is adult plasminogen. Thus neonates have an overall reduced capacity to respond to tPA in comparison with adults. If an infant’s condition does not respond to thrombolytic therapy, replacement of plasminogen should be considered.
Indications, Therapeutic Range, and Dose.
Infants in whom serious thrombotic complications develop, as defined by organ or limb impairment, may benefit from thrombolytic therapy. The clinical objective is removal of the clot as quickly and safely as possible. Surgical removal of a clot in a major vessel can be curative; however, it is technically difficult and poses a considerable life-threatening risk to infants, who are often premature. In the absence of contraindications, the use of thrombolytic agents in these infants is a preferred approach (see Chapter 33 ).
Conclusion
Over the past several decades, the developmental timing and functional maturation of the human hemostatic system have been examined in great detail. These studies show critically important differences in many components of the hemostatic system in premature and full-term newborns versus older children and adults. The reason for this unique physiologic state in the fetus and neonate is not clear but appears to be appropriately balanced for healthy full-term infants. Of clinical importance has been the determination of gestational and postnatal age-specific activity ranges of factors involved in hemostasis, which has allowed appropriate interpretation of clinical data. As newer diagnostic tests and treatments are introduced for older children and adults, it will be critical to carefully evaluate them separately in the context of neonates, both premature and full-term, given their unique physiologic state.
Acknowledgment
The author would like to acknowledge Dr. Paul Monagle and the late Dr. Maureen Andrew for their chapter on developmental hemostasis contained in previous editions of this textbook. Their work formed the basis for the current chapter. The author would also like to thank Dr. Ellis Neufeld, Dr. Marilyn Manco-Johnson, Dr. Martha Sola-Visner, and Dr. Bob Montgomery for helpful advice.
Appendix Normal Ranges of Hemostasis Laboratory Tests for Term and Preterm Infants
| FETUSES (WEEKS’ GESTATION) | |||||
|---|---|---|---|---|---|
| Parameter | 19-23 ( n = 20) | 24-29 ( n = 22) | 30-38 ( n = 22) | Newborn ( n = 60) | Adult ( n = 40) |
| PT (seconds) | 32.5 (19-45) | 32.2 (19-44) † | 22.6 (16-30) † | 16.7 (12-23.5) ‡ | 13.5 (11.4-14) |
| PT (INR) | 6.4 (1.7-11.1) | 6.2 (2.1-10.6) † | 3 (1.5-5) ‡ | 1.7 (0.9-2.7) ‡ | 1.1 (0.8-1.2) |
| APTT (seconds) | 168.8 (83-250) | 154 (87-210) † | 104.8 (76-128) † | 44.3 (35-52) ‡ | 33 (25-39) |
| TCT (seconds) | 34.2 (24-44) ‡ | 26.2 (24-28) | 21.4 (17-23.3) | 20.4 (15.2-25) † | 14 (12-16) |
| Factor I von Clauss (g/L) | 0.85 (0.57-1.50) | 1.12 (0.65-1.65) | 1.35 (1.25-1.65) | 1.68 (0.95-2.45) † | 3 (1.78-4.50) |
| Factor I antigen (g/L) | 1.08 (0.75-1.50) | 1.93 (1.56-2.40) | 1.94 (1.30-2.40) | 2.65 (1.68-3.60) † | 3.5 (2.50-5.20) |
| Factor IIc (%) | 16.9 (10-24) | 19.9 (11-30) ‡ | 27.9 (15-50) † | 43.5 (27-64) † | 98.7 (70-125) |
| Factor VIIc (%) | 27.4 (17-37) | 33.8 (18-48) ‡ | 45.9 (31-62) | 52.5 (28-78) † | 101.3 (68-130) |
| Factor IXc (%) | 10.1 (6-14) | 9.9 (5-15) | 12.3 (5-24) † | 31.8 (15-50) † | 104.8 (70-142) |
| Factor Xc (%) | 20.5 (14-29) | 24.9 (16-35) | 28 (16-36) † | 39.6 (21-65) † | 99.2 (75-125) |
| Factor Vc (%) | 32.1 (21-44) | 36.8 (25-50) | 48.9 (23-70) † | 89.9 (50-140) | 99.8 (65-140) |
| Factor VIIIc (%) | 34.5 (18-50) | 35.5 (20-52) | 50.1 (27-78) † | 94.3 (38-150) | 101.8 (55-170) |
| Factor XIc (%) | 13.2 (8-19) | 12.1 (6-22) | 14.8 (6-26) † | 37.2 (13-62) † | 100.2 (70-135) |
| Factor XIIc (%) | 14.9 (6-25) | 22.7 (6-40) | 25.8 (11-50) † | 69.8 (25-105) † | 101.4 (65-144) |
| PK (%) | 12.8 (8-19) | 15.4 (8-26) | 18.1 (8-28) † | 35.4 (21-53) † | 99.8 (65-135) |
| HMWK (%) | 15.4 (10-22) | 19.3 (10-26) | 23.6 (12-34) † | 38.9 (28-53) † | 98.8 (68-135) |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


