Allogeneic stem cell transplantation currently is the only curative option for severe β-thalassemia and sickle cell disease. Human globin gene therapy with autotransplantation of transduced human hematopoietic stem cells is an exciting alternative approach to a potential cure. One patient with thalassemia has recently been reported to have clinical benefit after lentiviral human β-globin gene therapy. He has not required blood transfusions for almost 2 years. Most of the patient’s gene correction and new human β-globin gene expression is caused by the expansion of a single clone in which the corrective transgene is inserted into an Hmga2 gene.
Human globin gene therapy is an exciting approach to curing homozygous β-thalassemia (β-thalassemia, Cooley anemia) as well as sickle cell anemia. These diseases are particularly suitable for this approach because the specific genetic defects that cause them are known: sickle cell disease is caused by a point mutation in the human β-globin gene; most β-thalassemia mutations are also caused by single nucleotide changes, all of which lead to either decreased or absent normal β-globin protein. Human β-globin gene therapy with autologous modified stem cells has been envisioned for many years by patients, physicians, and scientists as a logical and ideal way to cure the disease. However, it is only recently that some limited success has been achieved.
The only cure for β-thalassemia (Cooley anemia) is allogeneic stem cell transplantation (ASCT), using stem cells from adult peripheral blood, bone marrow, or umbilical cord blood sources. ASCT is discussed in detail by Gaziev and Lucarelli elsewhere in this issue , as well as by Kanathezhath and Walters. ASCT is limited by immunologic differences between patients and potential donors; less than 30% of patients have suitable donors. A curative result occurs in the most eligible patients who fit the criteria for transplantation, most of whom are children. The potential development of graft-versus-host disease, a potentially life-threatening complication caused by immune reactions, has tempered the use of ASCT, especially when a completely compatible donor is not available.
There are 2 general approaches to providing normal β-globin function by gene therapy in these disorders: correction of the DNA defect in the β-globin gene by homologous recombination, or addition of a normal β-globin gene to the genome. Gene correction has the great advantage of maintaining the β-globin gene in its native chromosomal environment, and thus is the preferred gene therapy approach. However, homologous recombination occurs at too low a frequency at present to be useful for human globin gene therapy.
Gene addition has been used successfully in human gene therapy clinical trials, with viral vectors transferring and expressing corrective genes in human hematopoietic cells. So-called γ-retroviral vectors containing Moloney viral components have been used to cure patients with severe immune disorders such as subacute combined immunodeficiency (SCID) and adenosine deaminase deficiency. In these conditions, the gene-corrected lymphocytes are naturally selected for survival and expansion in preference to the patient’s own defective cells, and even low-level transduction (infection) and expression of the corrective gene results in immune reconstitution of the affected T lymphocyte compartment, and cure.
No such selection currently exists for gene-corrected hematopoietic stem cells (HSC) containing and expressing the human β-globin gene in sickle cell disease or β-thalassemia. Thus, high levels of normal β-globin transfer and expression are required to cure these diseases.
Globin gene therapy
The current approach to human gene therapy for thalassemia is theoretically simple, using autotransplantation. HSC are taken from the patient, a normal hemoglobin (Hb) gene is added to the cells outside the body, and the human β-globin gene–corrected cells are returned to the patient intravenously. They automatically home and engraft in the marrow.
Gene therapy for β-thalassemia has been believed to be feasible since 1972, when β-globin complementary DNA, (cDNA), a copy of globin messenger RNA, was described. Then, it was believed that the globin cDNA itself could be used as the source of the normal human β-globin gene sequences that could cure the disease. However, in the 1980s, it became clear that, in addition to the coding sequences present in globin cDNA, other important regulatory elements are required for successful and high-level human β-globin gene expression. These sequences include the intervening sequences within the gene, and regulatory sequences upstream and downstream of the human β-globin gene. In the late 1980s, Grosveld and colleagues described important regulatory sequences far from the β-globin gene itself, called the β locus control region (β LCR) that are necessary to provide high level of expression of the human β-globin gene. The β LCR provides position-independent high-level enhancement of globin expression, and its discovery was seminal in moving β-globin gene therapy forward.
Viruses as Vectors
Naked DNA can theoretically be used as the vector (or carrier) to transfer and express genes in human gene therapy, including those for human β-globin. However, viruses are much more efficient. Viruses are pieces of RNA or DNA wrapped in specialized viral proteins: after infecting cells, viruses use the host cell’s molecular machinery to encode specific viral proteins, and express and assemble the proteins into viruses. Specific viral proteins on the surface of the viruses allow them to enter cells. After infection and integration, the viral DNA directs the synthesis of more of viral proteins; more viral particles assemble and are eventually extruded to infect more cells.
After infection, certain classes of viruses, adenoviruses, and, to some extent, adeno-associated viruses, remain in the cytoplasm of cells; they do not enter the nucleus and do not integrate into chromosomal DNA. These viruses are not useful for human blood stem cell gene therapy, a process that requires the corrective genes to integrate into the patient’s chromosomes; this is necessary so that, when the gene-corrected stem cells divide, the corrective genes are transferred as part of the chromosomal material and maintained in daughter stem cells.
RNA viruses, called retroviruses, are most useful for human gene therapy. These viruses contain so-called gag , pol , and env gene sequences that lead to the production of the proteins required by the virus: gag proteins produce the core proteins of the virus; the pol gene specifies the enzyme, reverse transcriptase, which the viruses use to make a DNA copy of their RNA; the env genes code for the proteins of the viral envelope. These retroviruses also have genes that encode a protein called integrase, which enhances the integration of viral genetic material into chromosomal DNA.
Intact replication-competent retroviruses produce more viral particles after integration, often kill the cells they infect, release their viral particles from the cells, and infect more cells. Replication-competent viruses are not desirable for human gene therapy; not only can they kill the cells they transduce, but their integration at multiple sites in host chromosomes can activate cellular oncogenes in a process known as insertional mutagenesis. Instead, in human gene therapy, we use pieces of viruses, not intact viruses, in such a way that they are incapable of generating intact viral copies of themselves, while still carrying genes, such as the human β-globin gene, into cells and integrating those genes into chromosomal DNA. We make so-called replication-incompetent defective retroviruses (pseudoviruses) that, unlike their normal counterparts, are unable to reproduce themselves after they have inserted their genetic material into our chromosomes.
These defective viruses do not contain all of the proper genes and signals for new wild-type viral production on a single piece of DNA or RNA, as in replication-competent viruses. Defective viruses are created in so-called packaging cells; these are tissue culture cells into which the genes that produce the necessary viral proteins, gag, pol, env, and integrase, usually derived from Moloney leukemia viruses, are added, The viral genes encoding these proteins are added to the packaging cells on separate pieces of DNA called plasmids. The production of viral proteins in the packaging cells leads to the formation of empty viral particles with no DNA or RNA material capable of chromosomal integration.
When a suitable piece of DNA containing the corrective gene (in our case a human β-globin gene–containing gene vector) is added to these packaging cells, so-called producer cells are made. The nucleotide sequences on this vector plasmid are the only ones that are integrated into the host chromosomes after viral integration. With the production of gag, pol, env, and integrase proteins in the producer cells, retroviral particles are formed containing the RNA encoding the corrective gene. The producer cells then release intact viral particles into the medium. The pseudoviruses containing the human β-globin gene transfer their globin gene sequences into target HSC, integrate the gene sequences into chromosomal DNA, and allow the expression of potentially curative human β-globin. To reiterate, these defective viruses, unlike their normal counterparts, cannot reproduce themselves after they have inserted their genetic material into our chromosomes; components of the material necessary to produce intact viruses in this gene therapy system are on separate plasmids and cannot generate normal infectious wild-type virus.
I believed many years ago that defective viruses containing the human β-globin gene could be used as a pill that could be taken orally to cure sickle cell disease and thalassemia. I believed that the pill would uncoat in the stomach; viral particles would be released and enter the blood stream, exit at the right tissue location (in this case, the bone marrow), and integrate into HSC DNA. Several tissue-specific viruses are known. Two examples are hepatitis virus, which contains envelope proteins that target specific receptors on liver cells; and the acquired immune deficiency syndrome (AIDS) virus, which only infects T lymphocytes. However, it has not been possible to find viruses with envelopes that specifically target human HSC.
Instead, the approach has been to collect and concentrate HSC ex vivo, and use envelope proteins that can enter many different cell types, including HSC. HSC are obtained from either bone marrow or circulating blood, and our human β-globin gene–containing retrovirus is added outside the body; this provides a great advantage compared with other gene therapy applications in which the target organ cannot be removed. Exposure of the virus is limited to the gene-targeted blood cells and there is no danger of affecting non–blood cells. In addition, the ex vivo approach permits high ratios of virus to relevant HSC, levels that might not be possible if the gene therapy virus was given in vivo.
We used Moloney leukemia virus–based γ-retroviruses, to transfer and express human genes in 2 phase 1 human clinical trials to express a potential anticancer gene, the multiple drug resistance (MDR) gene, in human HSC. For these trials, we developed safe and efficient defective packaging cell lines to produce the defective retroviruses. We achieved the expression of the MDR gene in the bone marrow of patients, but at levels that were too low to be of clinical significance.
Mouse Models
During the past 20 years, several groups have shown transfer and expression of a human β-globin gene into mouse HSC, occasionally at high levels, with γ-retroviral vectors. In 1997, we reported one mouse that made 20% as much human β-globin as mouse β-globin. However, this was a rare, significant, positive result among many negative ones.
Trial and error has shown that efficient globin gene therapy is not reproducible using γ-retroviral vectors. This finding is primarily because the target cell, the HSC, is largely quiescent; it divides infrequently. γ-Retroviruses require cell division to move their contents from the cytoplasm to the nucleus of cells. They are inefficient at having the viral particles enter the nucleus of HSC and integrating into the chromosomes of these cells that only occasionally divide.
To solve the problem of targeting nondividing human HSC, a special type of retrovirus, a so-called lentivirus, a virus that does not require cell division to achieve HSC target cell integration, is being used. Lentiviruses traverse the cytoplasm of nondividing cells and, after reverse transcription, lentiviral particles can move from the cytoplasm to the nucleus of cells without cell division. Sadelain and Leboulch pioneered the use of lentiviruses in human globin gene therapy. Working independently, they showed that the anemia in mice with diseases resembling human homozygous β-thalassemia (Cooley anemia) and sickle cell disease can be alleviated significantly using lentiviruses containing a normal human β-globin gene. These studies are the impetus for the current human globin clinical trial in Paris and proposed trials.
Sadelain and Leboulch were responsible for an earlier critical contribution to the human β-globin gene therapy field. They had previously shown that it was necessary to remove certain specific nucleotide sequences from human β-globin IVS2 for the globin retroviral vector to be appropriately reverse transcribed and expressed in target HSC.
Human globin gene therapy
Human globin gene therapy is essentially autologous stem cell transplantation with gene transfer. HSC from blood or marrow are removed from the patient, transduced with the corrective human β-globin gene–containing lentivirus, and returned to the patient by vein ( Fig. 1 ). This method overcomes the limitations of allogeneic transplantation because there are no immunologic barriers to engraftment, and, thus, many more patients are potentially eligible for treatment.
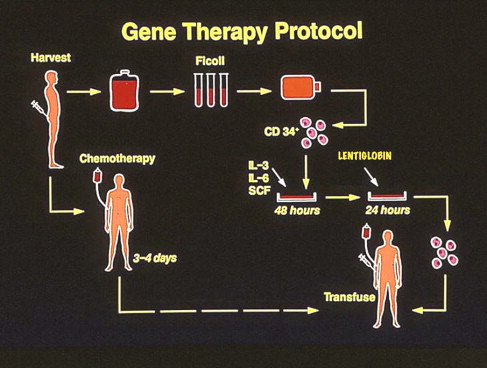
It is critical that human globin gene therapy specifically targets HSC, because these are the only cells capable of both cell division and HSC maintenance, as well as of erythroid differentiation. More differentiated transduced cells in the red blood cell lineage will merely continue to differentiate for days, lose their nuclei and become reticulocytes and mature red blood cells, and die.
In the past 2 decades, the environment for gene therapy research has been more favorable in France than in the United States. The unexpected death of a patient in a gene therapy trial at the University of Pennsylvania in 1999, using another type of virus, an adenovirus, had made this area of research less appealing to the scientific and medical community. The first largely successful human clinical gene therapy trial was performed using γ-retroviruses in Paris in the late 1990s by Cavazzana-Calvo and colleagues. Nine of the 10 children with X-linked SCID (X-SCID) in this trial were cured of the immunologic deficiency.
Current Human Thalassemia Clinical Trial
The first and only ongoing human β-globin gene therapy clinical trial, is being performed in Paris. A corrective human β-globin gene is being delivered using a proprietary lentiviral product called Lentiglobin (Genetics Pharmaceuticals Inc., Cambridge, MA). (I am a cofounder of Genetix, the primary medical consultant to it in the United States, and an equity shareholder). Lentiglobin contains the structural human β-globin gene as well as its promoter and critical elements of the human βLCR. Only these human β-globin gene–specific promoter-enhancer elements are actively transcribed after Lentiglobin sequences are integrated in the genome of gene-transduced target human HSC.
The Lentiglobin gene–containing plasmid also contains the human immunodeficiency virus (HIV) Rev response element and other DNA elements that enhance nuclear migration of the viral DNA. The Lentiglobin plasmid is a so-called self-inactivating (SIN) vector. It contains the 5′ HIV long terminal repeat (LTR) with its promoter and enhancer elements necessary for the transcription of Lentiglobin RNA and for the reverse transcription of this RNA as a DNA copy. However, the 3′ HIV LTR promoter–enhancer sequences in the SIN vector are rendered inactive by deletions. As in all retroviruses, when reverse transcription occurs, the 3′ HIV LTR becomes the 5′ LTR sequences; in our case, the resulting Lentiglobin DNA 5′ LTR sequences are, thus, rendered inactive. This Lentiglobin SIN vector structure ensures that no HIV promoter or enhancer sequences are active in transcription after integration of Lentiglobin DNA into the host cell genome.
In addition, the Lentiglobin vector expresses so-called insulator sequences, short DNA sequences placed on either side of the β-globin sequences in the Lentiglobin plasmid, which prevent the activation of genes upstream or downstream of the insertion sites of the Lentiglobin vector. The cells that produce Lentiglobin virus also contain separate plasmids that encode and express lentiviral HIV gag and pol genes, and the envelope protein, vesicular stomatitis viral protein (VSV-G). The Lentiglobin virus used in the trial has been shown to express human β-globin at high levels in target human HSC in mice.
One disadvantage of currently used lentiviral production, including Lentiglobin in the Paris trial, is that the expression of the viral HIV gag and pol proteins required are toxic to the producer cells. This toxicity requires that the Lentiglobin vector plasmid is transfected into a so-called transient packaging cell line system. In this system, the HIV gag-pol, VSV-G env, and Lentiglobin plasmids are added to the packaging cells, 293T monkey kidney cells, on separate plasmids, in small tissue culture dishes for 24 to 48 hours to transiently produce the Lentiglobin virus. Supernatants from several different tissue culture dishes are harvested and constitute the infectious Lentiglobin virus used in the trial. Pooled supernatants from the Lentiglobin virus–producing 293T cells are further purified by chromatography before being used to transduce the HSC target cells.
The use of transient lentiviral systems is not as convenient as more desirable stable producer lines developed with Moloney γ-retroviral components, in which all of the components of the virus are stably integrated into the packaging and producer cell lines; these stable lines were used in the human MDR gene therapy trials. The lack of toxicity of Moloney gag-pol expression permits single clones of γ-retroviral stable producer lines to be isolated and grown to large volumes without cell death and used repeatedly. Several potentially useful stable lentiviral packaging lines have been described recently in which the HIV gag and pol genes are stably integrated into chromosomal DNA. In these lines, the toxicity of the HIV gag-pol genes is circumvented by the transient and reversible expression of these genes using tetracycline control elements. However, none of these lines is currently used in human clinical trials.
In the Paris gene therapy trial, CD34+ cells are isolated from the bone marrow or mobilized peripheral blood samples of patients; this CD34+ cell purification eliminates many differentiated cells, and concentrates the HSC. As previously noted, human HSC are the only relevant population for human globin gene therapy because only HSC can both self-renew and differentiate into erythroid cells, presumably for the life of the patient. More differentiated nucleated erythroid cells differentiate and die in days to weeks.
A Unique Human β-Globin Gene–containing Vector
In the ongoing phase 1 clinical trial in Paris, the human β-globin gene sequence in Lentiglobin has been modified by mutating a single amino acid at position 87 of the β-globin sequence. Hb containing β87 globin (Hbβ87) has been shown to function normally in its expression and oxygen-carrying capacity. It has also been shown that Hbβ87 acts like fetal Hb (HbF) in preferentially interfering with sickling in studies of human sickled cells, and may be particularly useful in gene therapy trials in patients with sickle cell disease.
The major reason for using the β87 globin gene in Lentiglobin in patients with β-thalassemia is that Hbβ87 expression can easily be distinguished from that of normal Hb (HbA). Because all patients with thalassemia continue to have significant amounts of HbA in their blood as a result of transfusions after their transplantation, this distinction is extremely useful.
The amount of new β87 globin gene in the patients’ cells can be measured by polymerase chain reaction (PCR). New β87 globin protein is quantitated by high-pressure liquid chromatography (HPLC). Thus, positive tests for the presence of the β87 gene by PCR, and for β87 globin by HPLC, are clear measures of the success of human β87 globin gene transfer and expression in the trial.
In the Paris trial, bone marrow is harvested from patients, CD34+ cells isolated, and Lentiglobin is transduced into these cells (see Fig. 1 ). If adequate Lentiglobin transduction is documented in burst-forming unit, erythrocytes (BFU-E), and colony-forming units-granulocyte macrophage (CFU-GM) cultures from the patient’s transduced marrow samples, the patient then undergoes full myeloablation, and the transduced cells are returned to the patient intravenously (see Fig. 1 ). The current principal investigators (PIs) in the trial are Drs Leboulch and Marina Cavazzana-Calvo. Dr Eliane Gluckman was an earlier co-PI.
Clinical trial protocols are a compromise between potential benefit and perceived or known risk: the so-called benefit/risk ratio. In the Paris trial, we accepted the increased risk of full marrow ablation to increase our chances for meaningful therapeutic benefit. This risk was accepted because previous experience in human allogeneic bone marrow transplantation (ABMT) has shown that less-than-complete ablation of a recipient’s bone marrow is often insufficient to allow successful transplantation of donor cells. It has been shown in mice that HSC transduced with retroviruses compete unfavorably with wild-type HSC for marrow engraftment. This result suggests that, in human gene therapy protocols using reduced-intensity marrow ablation, the patient’s residual unmodified HSC will outcompete transduced gene–corrected cells for marrow engraftment.
However, the risks of complete marrow ablation are also potentially greater than using nonmyeloablative regimens, with longer periods of leukopenia and thrombocytopenia after transplantation.
Results to Date
Two patients with thalassemia have been treated to date on the current Paris protocol. The first patient, TK, is a woman with severe thalassemia. After the gene therapy procedure, she initially had evidence of a small amount of β87 globin gene transfer by PCR that was transient and too low to be clinically significant. However, her gene therapy treatment was not ideal because she received only one-third as many gene-corrected cells as planned. She survived several weeks of very low white blood cell and platelet counts, and was given antibiotics, white cell growth factors, and platelet transfusions to ameliorate these complications. She also continued to receive red blood cell transfusions to combat her anemia. She was eventually given an untransduced backup bone marrow sample (collected and stored as part of the protocol in case of the failure of the gene-corrected cells to engraft sufficiently). She has now fully recovered her white cell and platelet function.
The second patient, PLB, is a 19-year-old man, doubly heterozygous for β-thalassemia and HbE (α 2 β E 2 ). The β E gene acts like a β + thalassemia allele and only provides limited output of β E globin. The patient’s HbE gene output has always been low (HbE<20% of normal HbA levels), and his limited HbE and HbF production have not prevented him from severe lifelong anemia and its complications. He required monthly transfusions since early childhood before the gene therapy procedure. He also continued to need transfusions for several months after transplantation with the β87-containing Lentiglobin vector. In contrast to patient TK, he had only a short time after the gene therapy procedure of marrow hypoplasia during which his white blood cell (WBC) count and platelet count were low.
Several months after transplantation, PLB began to significantly increase his production of Hb β87. This increased production has continued in the last 19 months (to June 2010), and he has not needed any blood transfusions during this time. He is the first patient with a human Hb disorder to obtain clinical benefit and become transfusion independent with human β-globin gene therapy. He currently has approximately 9 to 10 g percent of Hb in his circulating blood, approximately one-third being Hb β87, one-third human HbF, and one-third HbE. He has no residual transfused HbA.
Safety Issues
During the past year, analysis of the clonal composition of patient PLBs reconstituting β87-containing cells by linear amplification-mediated PCR has revealed that, although reconstitution is polyclonal, a single clone is dominant. This clone has arisen from the insertion of the Lentiglobin vector DNA into a specific gene, Hmga2 ; expression of Hb β87 by this clone in erythroid cells is largely responsible for the Hb β87 production in the patient. Approximately 10% of the patient’s CFU-GM and WBCs as well as BFU-E contain the same insertion of Lentiglobin into Hmga2 , indicating that the clone is a multilineage clone, resembling a myeloid-biased HSC clone.
Insertion of Lentiglobin into one of the introns of Hmga2 with a loss of intronic function has occurred in this clone. This loss is similar to that occurring with other Hmga2 mutations, some of which are associated with clonal proliferation. Recent studies have shown that clonality, with loss of Hmga2 introns, is more common than was previously believed in patients in other human gene therapy trials ; in most of these trials, these clones, presumably selected for their increased proliferative capacity, do not continue to expand over time. The proliferation of the Lentiglobin-containing HMGA2 clone in PLB has continued at a low level in the past year. It is unknown whether the clone will expand, regress, or remain stable in the future.
The goals of human globin gene therapy are safety as well as efficacy. Experience from X-SCID trials is disconcerting in this regard. In these studies, although 9 of 10 patients in the Paris trial, and others in a similar trial in London, were cured of their disease, several patients have subsequently developed leukemia as a result of insertional mutagenesis. The unacceptable events in these trials are probably related, at least in part, to the insertion of the curative gene in an HSC in the vicinity of an oncogene, a gene whose expression can cause the uncontrolled growth of cells and cancer.
In X-SCID, normal T lymphocytes lack a normal γC receptor gene, whose protein product is necessary to fight certain types of infections. The curative γC receptor gene used in these gene therapy trials is expressed from a γ-retroviral promoter-enhancer LTR. In patients who developed leukemia, a rare cell inserted the γC receptor gene and its powerful nonspecific promoter-enhancer near a known oncogene, Lmo2 , and activation of Lmo2 likely caused the emergence of the proliferative leukemic clone.
The design of the Lentiglobin vector used in the Paris study theoretically avoids the potential problems of the γC vector in the X-SCID trial. As mentioned previously, first and most importantly, viral enhancer elements are not activated in the SIN Lentiglobin vector, as with the γC vector. Instead, only human β-globin gene–specific promoters and enhancers direct human β-globin gene expression, and these are only active in red blood cells. In addition, DNA sequences called insulators have been added to the Lentiglobin vector to prevent any potential activation of oncogene sequences, and the insertional mutagenesis seen in the X-SCID trial.
In patient PLB, the activation of HMGA2-mediated proliferation of a clone of HSC-like cells was not caused by activation of oncogenes by retroviral elements. The proliferative clone is most likely the effect of the insertion of the Lentiglobin vector directly into the intron of Hmga2 in one of the marrow stem cells during the initial transduction of the patient’s HSC. Subsequently, the mutated clone was presumably selected for proliferation; increased HMGA2 protein expression by the clone in PLB’s cells has been confirmed in laboratory studies.
Other Lentiglobin Human Gene Therapy Trials
Additional patients with severe thalassemia are currently being recruited to the Paris trial. In addition, Dr Sadelain and his associates have planned a clinical trial with autotransplantation to begin in the United States in the near future. This trial will use a lentiviral human globin gene–containing vector designed by Dr Sadelain that is somewhat different from Lentiglobin.
The only other human gene therapy trial using lentiviral vectors, other than the human globin gene trial, has been reported recently in patients with the neurologic disease, X-linked adrenoleukeukodystrophy (ALD). This is a rare disorder caused by a genetic defect in the ABCD1 receptor gene in which there is deficiency of ALD protein, an ATP-binding cassette transporter protein, required for normal myelination of neurons. The disease is progressive and must be treated early in life to prevent severe neurologic disability.
ALD can be cured by ASCT. Macrophages produced by HSC have been shown to migrate to the central nervous system (CNS), to become functional glial cells capable of producing the normal required protein. In the gene therapy clinical trial of patients with ALD, also performed in Paris, 3 boys with ALD deficiency were treated. CD34+ cells were transduced with an ALD gene–containing lentiviral vector using procedures similar to those used in the Paris human globin gene trial. In all 3 treated ALD patients, there was a lack of progression of CNS demyelination compared with that in untreated patients with ALD. The results are encouraging and comparable with the relative clinical benefit obtained using ASCT. To date, there has been no evidence of specific clonal proliferation in any of the 3 ALD lentiviral vector–treated patients.
A New Homologous Recombination Approach to Thalassemia Gene Therapy
As discussed earlier, ideal human globin gene therapy for Cooley anemia as well as sickle cell anemia would be gene correction: the correction of the single base mutation in the DNA of the mutated human β-globin gene at its normal chromosomal position. This correction can be accomplished by adding a vast excess of DNA containing the normal β-globin DNA sequence to correct the mutant DNA sequence by homologous recombination in the HSC of patients with sickle cell disease and thalassemia. Gene correction has a great advantage compared with gene addition in that there is no possibility of insertional mutagenesis because the corrective piece of DNA used is short, biologically inert, and contains no viral elements; the only change in the patient’s chromosomes is that the mutant β-globin gene is corrected.
However, as mentioned earlier, the frequency of gene correction occurring using HSC is currently too low to be clinically useful. This shortcoming is primarily because human HSC are limited in number and cannot be grown to large amounts in culture: they differentiate preferentially into later blood cell elements rather than dividing and reproducing themselves in large enough numbers to be useful for gene therapy.
Homologous recombination with gene correction has been successfully used for many years to correct several gene defects in mice using embryonic stem (ES) cells, the multipotential cells that are capable of producing any tissue of the animal. This is because, in contrast with HSC, ES cells can be grown to large numbers without altering their biologic properties. These large numbers of cells are required for gene correction because it is such a rare event, and many individual cells must be isolated and analyzed before sufficient gene-corrected cells can be obtained.
However, in the past 5 years 2 extraordinary advances have occurred that increase the possibility that a gene correction strategy will eventually be used for gene therapy for thalassemia and sickle cell disease. First, Takahashi and Yamanaka and Takahashi and colleagues in Japan showed that human as well as mouse ES cells can be derived by manipulating skin cells from each of these sources. In these experiments, the addition and expression of just 4 genes ( Klf4 , Oct4 , Sox2 , and c-Myc ) rewire the circuitry of the differentiated skin cells so that they acquire many of the characteristics of true ES cells. These reprogrammed cells are called induced pluripotent stem (iPS) cells. Before this discovery, human ES cells could only be obtained from living human embryos; this has been considered unethical by religious groups because it involves the destruction of embryos. The use of the patients’ own skin cells to obtain ES cells greatly diminishes these concerns.
Second, it has been shown that mouse iPS cells derived from skin cells and manipulated in tissue culture can be used to cure mice with the equivalent of human sickle cell anemia. In these experiments, skin cells from these mice were converted to iPS by the addition of the 4 special genes mentioned earlier; short inert pieces of normal human β-globin sequence DNA, containing sequences that correct the sickle cell mutation, were added to the skin-derived iPS of these sickle mice; iPS were grown to large amounts and the rare iPS in which the sickle cell mutation was corrected were isolated; the globin gene–corrected iPS were grown to large amounts and, after being treated in cell culture with growth factors and chemicals, became HSC. These human β-globin gene–corrected HSC, originating from skin cells of mice, were then used to reconstitute the ablated bone marrow of the sickle mice and largely cured their sickle cell anemia, as assessed by correction of anemia and production of normal HbA.
Using an iPS strategy, as with our Lentiglobin gene therapy, there are no immunologic barriers to transplantation because the skin cells originally used are derived from the patient. However, as previously noted, a great advantage of this skin to ES cell approach compared with our Lentiglobin addition gene therapy approach is that it avoids the possibility of insertional mutagenesis because no functional DNA is added to the patient’s chromosomes.
One of the genes initially used to convert skin fibroblasts to iPS cells was c-myc , a known oncogene; increased expression of its protein product, c-Myc, is associated with cancer. More recently, several advances, including protocols without increased c-myc , have been made to address the problem. Such safer methods of generating iPS cells from skin cells, avoiding c-Myc expression, and using small molecules and/or the required proteins themselves rather than retroviral vectors producing those proteins, may eventually make the use of human iPS cells more feasible for human β-globin gene therapy. It will also be necessary to develop efficient methods for the conversion of human iPS cells to human HSC to accomplish this goal.
Other Approaches to Thalassemia Gene Therapy
Another approach to curing β-thalassemia is to increase human HbF (α 2 γ 2 ) production. Studies leading to clinical trials using a human γ-globin–containing vector, instead of a human β-globin vector, have been proposed.
Recent success in understanding γ-globin gene regulation and human γ- to β-globin switching in late fetal life has also suggested a different approach to thalassemia and sickle cell gene therapy. Earlier, it had been shown that the protein Ikaros was an important regulator of human γ- to β-globin switching from data in mouse models. More recently, it has been found that the action of a single human gene, BCL11A , is much more potent than that of Ikaros .
Like Ikaros, BCL11A is expressed primarily in adult-type hematopoietic cells and forms chromatin remodeling complexes in these cells that suppress γ-globin production. In mice containing human γ- and β-globin transgenes, it has recently been shown that deletion of BCL11A leads to continued high-level production of HbF. If an antisense strategy can be found to prevent BCL11A action in adult HSC, then continued high-level HbF production might cure β-thalassemia and sickle cell disease. Recent oligonucleotide and antisense RNA strategies may be useful in inhibiting BCL11A expression in human HSC, and provide the basis for such a new globin gene therapy approach.
Strategies for the selection and enrichment of HSC containing and expressing a curative human β-globin gene have been of interest for the past 2 decades. In this scenario, a retroviral vector plasmid containing both a selectable gene and a human β-globin gene is used. The selectable gene, most commonly the human MDR or methylguanine transferase ( MGMT ) gene, is one that is normally expressed at low levels in human HSC. Transfer and expression of retroviral vectors that permit high-level expression of MDR or MGMT in HSC then allow the preferential survival of transduced cells, after systemic administration of certain drugs that kill cells expressing only low levels of these proteins. Using this strategy, mouse HSC containing the MGMT and MDR vectors alone have been shown to be preferentially selected by drug administration in intact animals.
However, human clinical trials using the human MDR or MGMT genes alone for HSC selection have been unsuccessful in significantly enriching for gene-expressing cells. Most recently, a lentiviral vector containing both the human γ-globin gene and the MGMT gene has been shown to select for cells containing and expressing this bicistronic vector in mice.
Related posts:
 The Population Genetics and Dynamics of the Thalassemias
Noninvasive Approaches to Prenatal Diagnosis of Hemoglobinopathies Using Fetal DNA in Maternal Plasma
Pharmacologic Induction of Fetal Hemoglobin Production
Thalassemia: An Overview of 50 Years of Clinical Research
Pharmacologic Induction of Fetal Hemoglobin Production
Allogeneic Cellular Gene Therapy for Hemoglobinopathies
The Population Genetics and Dynamics of the Thalassemias
Noninvasive Approaches to Prenatal Diagnosis of Hemoglobinopathies Using Fetal DNA in Maternal Plasma
Pharmacologic Induction of Fetal Hemoglobin Production
Thalassemia: An Overview of 50 Years of Clinical Research
Pharmacologic Induction of Fetal Hemoglobin Production
Allogeneic Cellular Gene Therapy for Hemoglobinopathies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


