Fig. 13.1
Frequency of specific late effects in survivors of childhood cancer treated with or without HSCT and their siblings (adapted from Armenian et al. [2])
Further, survivors of HSCT were more likely to report adverse general health, functional impairment and activity limitations.
Survivors of HSCT are also at risk for early mortality, even if they survive beyond the peri-transplant period. The BMTSS reported that patients transplanted prior to age 18 years who survive for 2 years after their transplant are almost 17 times more likely to die than the similarly aged general population [3]. Although recurrence of the primary disease is the leading cause of death in HSCT survivors, cGVHD, infection, subsequent malignant neoplasms and specific organ toxicities (e.g. cardiac, pulmonary) can also cause premature death.
In this chapter, we will discuss the long-term outcomes observed after HSCT during childhood or adolescence, and will examine the impact of the type of transplantation, the conditioning regimen and the presence or absence of cGVHD on the risk for these outcomes. Given their risks, all survivors of childhood HSCT require lifelong follow-up care from a clinician with expertise in the late effects observed in these survivors and the surveillance and health promotion strategies needed to minimize long-term morbidity. We will present the health care requirements of childhood HSCT survivors and will summarize the existing surveillance recommendations for this growing population.
13.2 Types of HSCT
13.2.1 Autologous HSCT
Prior to the introduction of high-dose chemotherapy with autologous stem cell rescue (autologous HSCT), delayed or absent bone marrow recovery after high-dose chemotherapy limited the intensity of chemotherapy that could be given to children with some cancers. The development of technologies to harvest a patient’s stem cells, store them and subsequently re-infuse them has allowed for the safe delivery of doses of chemotherapy above bone marrow tolerance [4]. The potential for improved disease-free survival after autologous HSCT must be balanced with the increased risk of treatment-related morbidity associated with such intense regimens. Although there is a small increase in the risk for transplant-related mortality during the acute phase of autologous HSCT, the risk of late effects and the long-term follow-up required after this procedure are similar to those in children who have been treated with intensive chemotherapy regimens without autologous HSCT. After transplant, these children are usually followed by their primary oncology team rather than a stem cell transplant specialist.
13.2.2 Allogeneic HSCT
Over the last four decades, allogeneic HSCT has been established as a curative option for many children with malignant and non-malignant diseases. Advances in supportive care, donor and patient selection, and better design and delivery of conditioning regimens have led to improved patient outcomes. However, as more children survive after allogeneic HSCT, special attention must be paid to the development of late effects. The increasing availability of unrelated donors along with a worldwide unrelated cord progenitor stem cell banks allowing HSCT with one or more human leukocyte antigen (HLA) mismatches has led to better survival because of graft-versus-leukemia (GVL) effect, but at the expense of a significant increase in HSCT late effects due to cGVHD and its life-long complications. Compared to childhood cancer survivors in general, survivors of allogeneic HSCT have a higher incidence of late effects, with 93 % of children reporting at least one late effect at a median follow-up of 7 years after HSCT. In 25 % of these children, the late effects are severe, disabling or even life threatening [5]. This chapter will focus on the late effects that can arise from allogeneic HSCT conditioning regimens and from the subsequent development of GVHD, particularly the chronic form.
13.3 Graft-Versus-Host Disease (GVHD)
13.3.1 Acute GVHD
The availability of new stem cell sources and the development of non-myeloablative conditioning regimens that are associated with later onset GVHD have negated the historical definition of GVHD. Previously, acute GVHD (aGVHD) was diagnosed when the disease occurred before day 100 after HSCT, in contrast to chronic GVHD (cGVHD) which was defined as occurring beyond day 100 [6]. Although cGVHD is the main contributor to late effects in long-term survivors, there are occasions where aGVHD remains active years after HSCT. This may manifest as inflammatory dermatitis, enteritis or hepatitis with persistence of cytotoxic lymphocytes and inflammatory cytokines that can cause ongoing tissue and organ damage [7, 8]. Prolonged aGVHD and its treatment can lead to further immunosuppression and delayed immune recovery with a risk for repeated infections in long-term survivors. A salient example of an acute transplant-related process that can contribute to a chronic late effect is the “allo lung syndrome”. In this syndrome, a respiratory infection that occurs shortly after HSCT contributes significantly to the later development of permanent lung damage manifested as bronchiolitis obliterans (BO) in the context of cGVHD [9]. aGVHD has also been established as a major risk factor for the development of cGVHD with consequent late effects in HSCT survivors [10–14].
13.3.2 Chronic GVHD
Chronic GVHD (cGVHD) is the most important cause of non-relapse morbidity and mortality following allogeneic HSCT [15, 16]. Although the incidence of cGVHD is lower in children (30–50 %) than adults (70–80 %) [17], its prevalence has increased dramatically in recent years due to the use of stem cell sources other than bone marrow, particularly peripheral blood stem cells (PBSC) [18, 19]. In general, umbilical cord blood (UCB) transplantation is associated with a lower risk of cGVHD [20]; however, since greater HLA disparity is tolerated when choosing a UCB donor, more children may develop cGVHD after UCB transplantation.
The presence of cGVHD is directly related to decreases in quality of life, impaired mental health, activity limitations and impaired physical function, and inferior long-term survival after HSCT [21–23]. cGVHD grading systems have important prognostic significance. Historically, severity was characterized as either “limited” or “extensive” [24]. These definitions have been expanded to include other measurements that correlate with prognosis, namely thrombocytopenia, progression of GVHD despite therapy, extensive skin involvement, gastrointestinal involvement, and low performance status at diagnosis of cGVHD [25, 26]. Recently, the National Institutes of Health (NIH) published consensus criteria which classify cGVHD as mild, moderate or severe [6].
Figure 13.2 displays many of the manifestations of cGVHD.
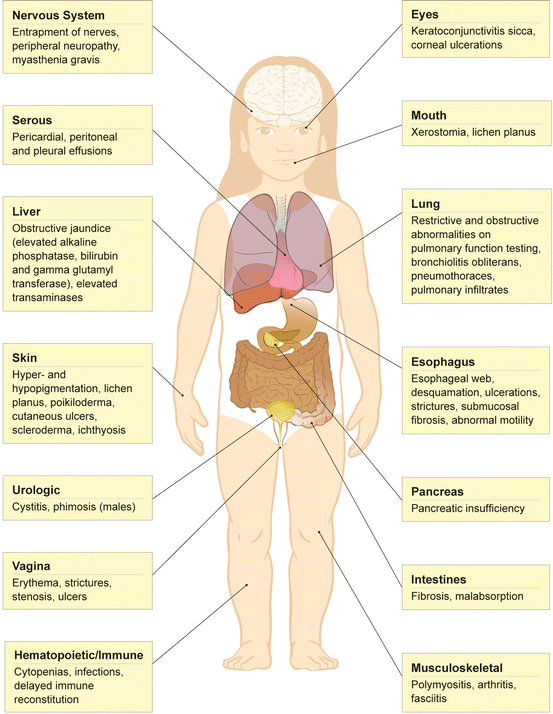
Fig. 13.2
Manifestations of chronic graft-versus-host disease in survivors of allogeneic HSCT
Treatment of cGVHD in children varies between transplant centers and is generally derived from the adult experience. Despite there being no proven standard therapy for cGVHD, cyclosporine and prednisone are commonly used as frontline treatment. Therapy may be needed for a long period of time (5 years or longer) [27]. Initial response to therapy is vital, as there is strong evidence that 90 % of patients who will ultimately respond to therapy will have shown signs of improvement by 3 months after initiating treatment. Patients should be observed carefully for response to frontline therapy, GVHD-related complications and morbidities stemming from glucocorticoid and immunosuppressant therapy. These include recurrent infections which can be bacterial, viral or fungal. Such infections may be life threatening and vigilance is required to prevent or promptly treat infections even years after the HSCT. Other side effects such as hypertension, diabetes and new malignancies can occur after prolonged immunosuppressant therapy. If frontline treatment is unsuccessful, salvage regimens include sirolimus, mycophenolate mofetil (MMF), pentostatin, rituximab, extracorporeal photopheresis (ECP) and targeted agents such as daclizumab and etanercept. These agents are potent immunosuppressants. ECP may increase the risk for infections because of a prolonged need for a large central venous access. These catheters may also increase the risk of thromboses and can hinder daily activities and life style. Furthermore, therapy for cGVHD is usually needed for a prolonged period, and clinicians must consider the impact of therapy on growth (especially with prolonged use of glucocorticoids), nutrition, organ function (in particular, the impact of the calcineurin inhibitors such as cyclosporine, tacrolimus and sirolimus on kidney function), immune reconstitution and psychosocial functioning.
13.4 Pulmonary Late Effects
Pulmonary toxicity is a major cause of both acute and long-term morbidity and mortality after HSCT [28]. Bronchiolitis obliterans (BO) is a well recognized cause of severe lung damage with a high rate of mortality [29–31]. The median time to BO onset after HSCT is 328 days (range 48–927 days) [32]. Risk factors for BO development include patient factors (such as reduced baseline respiratory status prior to HSCT), donor factors (such as related or unrelated donor, the degree of HLA matching and the stem cell source), and transplant factors (such as the intensity of the conditioning regimen and the type of GVHD prophylaxis). cGVHD is the most important risk factor [33–38]. Although establishing a diagnosis of BO requires the exclusion of intercurrent infection, there is increasing evidence that respiratory tract infections early after HSCT increase the risk of developing BO at a later date (the “allo lung syndrome”, discussed above) [9]. Pathologically, BO results from obstruction and/or obliteration of the small airways due to luminal occlusion of the terminal and respiratory bronchioles caused by inflammation and fibrosis. In severe cases, the airway develops circumferential scarring with complete obliteration of the lumen leading to the clinical presentation of obstructive lung disease (OLD) characterized by cough, shortness of breath and/or wheezing [39, 40].
Restrictive lung disease (RLD) and impaired diffusion capacity are more common than OLD. They may represent persistence of lung injury sustained prior to HSCT, usually as a result of previous treatment with chemotherapy or radiation [41, 42]. The risk for RLD is modified by the transplant conditioning regimen, the primary cancer diagnosis, the development of scleroderma or contractures, and donor relation. Patients treated with Busulfan or total body irradiation (TBI) have the highest risk of RLD. In the largest pediatric study published to date, pulmonary function tests were performed at a median of 10 years (range 5.0–27.5) after HSCT [43]. Forty percent of survivors had either RLD or OLD, and at least 15 % had an isolated low diffusion capacity of lung for carbon monoxide (DLCO). Moderate-to-severe pulmonary function impairment was present in 45 % of patients with RLD or OLD. In addition to RLD and OLD, other lung morbidities after transplant include recurrent lower respiratory infections and chest wall fibrosis.
Prior to the development of irreversible lung damage, children may have a prolonged asymptomatic period or symptoms that are attributed to infections. Survivors may present with subtle chest symptoms such as a dry cough. Progressive shortness of breath and wheezing might develop over time. In severe cases, tachypnea and weight loss due to increased work of breathing may ensue. If there is a suggestion of pulmonary disease on a survivor’s routine history or physical examination then a chest X-ray or high resolution CT scan may reveal an infectious process or air trapping. Pneumothorax, pneumomediastinum, and subcutaneous emphysema are usually signs of advanced disease. Serial pulmonary function testing should be performed in any survivor at risk for pulmonary disease since the NIH guidelines advise that a diagnosis of BO may be suggested based on these tests alone [6]. All survivors of allogeneic HSCT who are able to perform PFTs should have regular testing every 6 months in the first 2 years after HSCT and less frequently thereafter if they are asymptomatic and their PFTs are stable. Consideration for more frequent PFT screening in recipients of mismatched unrelated donor grafts, or patients with active cGVHD is advised. Invasive procedures such as bronchoalveolar and lung biopsy are reserved for survivors with significant pulmonary disease or when the diagnosis is unclear. Early diagnosis is crucial to preserving pulmonary function with the use of effective immunosuppressant agents such as high-dose systemic pulse glucocorticoids which may stabilize OLD and prevent further progression [44]. Other treatment modalities are mainly supportive, and include inhaled glucocorticoids, bronchodilators, oxygen support, and pulmonary rehabilitation. Lung transplantation has been performed on rare occasions when there is an appropriate candidate with severe BO. Given the complexity of lung complications after HSCT, treatment of BO and other pulmonary morbidities is usually a co-ordinated effort between the transplant team and a respiratory physician with expertise in lung injury after HSCT.
13.5 Gastrointestinal Tract and Hepatic Late Effects
Late effects involving the gastrointestinal (GI) tract occur mainly as a result of cGVHD. These late effects may also represent persistence of GI pathology that arises during the acute transplant period. Symptoms include nausea, loss of appetite, non-specific abdominal pain, weight loss, cramping or diarrhea [45]. In severe cases of cGVHD, intestinal malabsorption and pancreatic insufficiency may lead to weight loss, and trace element and vitamin deficiencies [46]. Rarely, esophageal stricture resembling achalasia occurs with resulting dysphagia. Unexplained peritoneal effusion has also been reported [47].
The most common long-term hepatic presentation after HSCT is a non-specific elevation of liver enzymes with normal synthetic and metabolic function and no signs or symptoms of liver disease [48, 49]. In most cases, no particular cause is found. However, causes of abnormal liver enzymes after HSCT include cGVHD, iron overload, chronic viral hepatitis B or C, autoimmune hepatitis and prolonged total parenteral nutrition (TPN) [50–55]. cGVHD involving the liver usually manifests with cholestasis and obstructive jaundice as a result of fibrosis—patients present with increased alkaline phosphatase (ALP), gamma glutamyl transferase (GGT) and serum bilirubin levels [56]. Recently, hepatic cGVHD has been recognized more frequently in children with isolated increases in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) [57]. The development of focal nodular hyperplasia (FNH) is also reported after HSCT. This benign liver tumor may mimic metastases or subsequent malignant neoplasms [58].
Physicians caring for HSCT survivors must obtain a detailed diet history, chart height and weight, and inquire as to symptoms of abdominal pain, diarrhea, nausea and vomiting. If there is concern about GI or liver dysfunction then the work-up should include assessment of hepatic function as well as testing for malabsorption or pancreatic insufficiency (serum trypsinogen, fecal fat assessment, and vitamin and trace element levels). In cases of dysphagia, a chest X-ray or a barium study may be needed to establish the diagnosis. Referral to a gastroenterologist may prompt upper or lower GI endoscopy or liver biopsy in certain cases.
13.5.1 Iron Overload Post HSCT
Iron overload is a common contributor to liver dysfunction after HSCT in children with thalassemia as well as in those transplanted for hematological malignancies [59, 60]. Abnormal liver function tests are common signs of iron overload after HSCT. However, liver function abnormalities can also be caused by GVHD, drug toxicity and infections [60]. In children with pre-existing iron overload, red cell transfusions during transplantation can further damage the liver and other organs. Children without pre-existing iron overload are at risk for the development of iron overload due to the multiple transfusions in the peri-transplant period. In a study of 13 children with normal abdominal MRI scans prior to HSCT, 10 (77 %) were noted to have post-transplant iron overload in the liver. Six showed iron overload in the spleen (46 %) and four in the bone marrow (38.5 %) [61]. Although the majority of liver biopsies carried out in the late post transplant period are performed to assess for GVHD, these biopsies commonly demonstrate significant iron overload [62]. MRI can be used to measure liver iron concentration non-invasively, and to delineate specific areas suitable to perform these measurements [63]. In contrast, estimates obtained by needle biopsy show considerable variability depending on site of biopsy [64]. Serum ferritin, although less specific, has been shown to be an important marker for iron overload and may be a valuable tool for monitoring response to phlebotomy or iron chelation [54].
13.6 Hematopoietic and Immune Dysfunction
Prolonged hematopoietic or immune system dysfunction occurs mainly in the context of GVHD. Damage to the bone marrow stroma or autoimmune phenomena can lead to cytopenias [59–61, 65–67]. Thrombocytopenia is the most common hematopoietic manifestation of cGVHD and usually confers an inferior outcome [25].
Immune reconstitution after HSCT is complex—both recipient and donor factors play a role. Important recipient factors include the patient’s age (with older children at a greater risk for delayed immune recovery, particularly after a UCB HSCT), the primary disease and its therapy, the patient’s general condition at time of HSCT, the conditioning regimen and GVHD prophylaxis [68]. The type and source of the graft substantially affect the recovery of the recipient’s immune function. For example, T-cell depleted and UCB grafts require more time for immune recovery. Natural killer (NK) cells are the first immune cells to reconstitute and may be present as early as 100 days after transplant. The B and T lymphocytes are reduced in numbers and function for many months after the HSCT. However, in the absence of significant GVHD, they gradually normalize after 1 year [69]. This process is much slower when UCB is used and may take up to 2 years for reasonable immune function [70, 71]. While engraftment and restoration of immune function is normally quicker after PBSC than after BM or UCB grafts, the higher incidence of GVHD in children treated with PBSC may compromise immune recovery. Generally, for all the three stem cell sources, the presence or absence of GVHD (particularly cGVHD) is the most important factor influencing immune reconstitution [72].
Children require repeated administration of vaccinations following recovery from allogeneic HSCT. The United States Centers for Disease Control (CDC) and the European Blood and Marrow Transplantation group (EBMT) have published recommendations about the timing of initiation of revaccination after HSCT [73]. Factors such as immunosuppressive therapy and the presence of GVHD may affect these timelines in individual patients. A recommended vaccine schedule is shown in Table 13.1.
Table 13.1
Suggested immunization schedule for children after allogeneic HSCT
Timing post HSCT | Diphtheria, tetanus and pertussisa | HiB | IPV | Pneumo-coccus | Men‐C | MMR | Var | HB | HPV | Influenza |
|---|---|---|---|---|---|---|---|---|---|---|
12 months | ■ | ■ | ■ | ■b | ■ | – | – | ■ | – | – |
14 months | ■ | ■ | ■ | ■ | – | – | – | ■ | – | – |
16 months | – | – | – | (■)c | – | – | – | – | – | – |
24 months | ■ | ■ | ■ | – | – | – | – | – | – | – |
24 months | – | – | – | ■ | – | ■ | ■d | ■ | – | – |
30–36 months | – | – | – | – | – | ■ | – | – | – | – |
4–6 years old | (■)e | – | – | – | – | – | – | – | – | – |
Grade 8 females | – | – | – | – | – | – | – | – | ■ | – |
14–16 years old | (■)f | – | – | – | – | – | – | – | – | – |
Every Autumn | – | – | – | – | – | – | – | – | – | ■g |
Surveillance for hematopoietic system recovery involves periodic complete blood counts and occasionally bone marrow testing if there are prolonged cytopenias. As many infections lead to myelosuppression, testing for infectious agents such as viruses is warranted. Immune reconstitution studies are the mainstay of immunologic surveillance after HSCT. While some transplant centers routinely perform lymphocyte subset analysis, lymphocyte proliferation studies and assess for antibody response to common vaccines, other centers will perform these tests in only selected patients such as those patients with cGVHD, or when a decision regarding immunization or antiviral therapy is dependent on these tests.
13.7 Cutaneous, Ocular, Dental, and Oral Late Effects
Cutaneous late effects usually occur in the context of cGVHD. Manifestations include hypo- or hyperpigmentation, lichen planus, poikiloderma, telangiectasias, cutaneous ulcers, sclerodermatous skin changes that may lead to decreased joint movements and contractures, ichthyosis, onychodystrophy, alopecia and loss of sweat glands. While superficial cGVHD involving the skin can lead to dry skin, pruritus, erythema and rash, deep sclerotic changes resulting from inflammation and fibrosis of the dermis, subcutaneous tissue, or fascia can lead to significant long-term functional disability [74]. Skin care should include attention to topical moisturizers, sun protection, and surveillance for skin cancers.
Cataracts are the most reported ocular late effects in children after HSCT. Incidence has been reported to be between 30 % and 75 % in different studies [75, 76]. The use of TBI is strongly associated with later development of cataracts [77, 78]. Prolonged use of glucocorticoids and the presence of cGVHD may also play a role in cataract development. Other ocular late effects, mainly related to cGVHD, include cicatricial conjunctivitis, keratoconjunctivitis sicca, punctate keratopathy and blepharitis. Symptoms include dry, gritty or painful eyes and photophobia leading to reduction in vision and in severe cases, blindness. Surveillance for cataracts is recommended for all survivors after HSCT, particularly those at highest risk (TBI recipients, prolonged corticosteroid use and cGVHD). Specialized ophthalmology care is recommended for long-term follow-up of ocular cGVHD to prevent irreversible damage.
Dental late effects after HSCT include abnormal development of teeth (tooth agenesis, hypodontia, microdontia and abnormal roots), delayed eruption, over-retention and increased risk for dental caries [79, 80]. Children younger than 5 years of age at the time of HSCT have the highest risk of dental late effects [79, 80]. Radiation involving the head and neck is a recognized risk factor for dental abnormalities [81, 82].
Oral late effects such as xerostomia, gingivitis, fibrosis and decreased salivary flow are more common in the context of cGVHD and can lead to significant reduction in quality of life [83]. These changes may increase the risk for secondary viral (especially herpes simplex) and yeast infections [84]. Furthermore, oral cGVHD and the topical immunosuppressant therapy used to control it may increase risk of subsequent malignant neoplasms such as oral squamous cell carcinoma [85]. Regular dental care by a dentist who is familiar with HSCT-related oral and dental complications and strict oral hygiene are vital in preventing or reducing these late effects.
13.8 Subsequent Malignant Neoplasms
Survivors of HSCT are at increased risk for the development of a subsequent malignant neoplasm (SMN). Their risk is increased if their pre-HSCT treatment includes radiation therapy or exposure to certain classes of chemotherapy agents, particularly oxazaphosphorines (e.g. cyclophosphamide) and epipodophyllotoxins (e.g. etoposide), or if their HSCT conditioning includes total body irradiation (TBI), or agents such as cyclophosphamide or etoposide. The risk is compounded by the development of cGVHD or the need for prolonged immunosuppression. HSCT survivors are at risk for solid neoplasms, hematological malignancies, and post transplant lymphoproliferative disorders (PTLD). The BMTSS reported that survivors of pediatric HSCT were almost 9 times as likely as survivors treated without HSCT to develop a SMN, even when controlling for exposure to brain or chest radiation and treatment with oxazaphosphorines or epipodophyllotoxins [86].
Solid neoplasms have been reported with increased frequency, particularly in survivors of allogeneic HSCT. The incidence of these malignancies increases with time, and does not appear to plateau [87]. Radiation therapy is likely the most important risk factor for their development, and several studies (but not all) have demonstrated that TBI conditioning increases the risk [88, 89]. Tumors of the thyroid as well as oral cancers are among the more common SMN observed [90, 91]. Non-melanoma skin cancers (basal cell carcinoma, squamous cell carcinoma) have also been observed with increased frequency in these patients [92].
Secondary leukemias and myelodysplastic syndrome (MDS) occur with increased frequency in survivors of autologous HSCT [93], but are uncommon after allogenic HSCT [87]. Pre-transplant therapy with oxazaphosphorines or epipodophyllotoxins and conditioning with TBI have been associated with increased risk. These malignancies are more common in adults treated with autologous HSCT than in similarly treated children [93].
Finally, post transplant lymphoproliferative disease (PTLD) is an uncommon occurrence after allogeneic HSCT. Risk factors include HLA disparity, T-cell depletion of donor marrow, use of anti-thymocyte globulin and both acute and chronic GVHD [93, 94]. Most cases are associated with Epstein-Barr Virus (EBV) infection [93]. PTLD usually develops in the first year after transplant, so it is infrequently a problem in long-term survivors. Survival is poor [91].
The Children’s Oncology Group’s (COG) Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers (available at www.survivorshipguidelines.org) present recommendations for surveillance of SMN in survivors deemed to be at increased risk. Survivors must also be encouraged to comply with cancer screening advocated for the general population (e.g. PAP smears, routine mammography, colorectal cancer screening) and to adopt lifestyle practices (such as sun protection and avoiding cigarette smoking) that will reduce their long-term cancer risks.
13.9 Endocrine Late Effects
Among the many late effects of childhood cancer therapy and HSCT, endocrine manifestations are particularly common. Complications derive from damage to endocrine tissues as a result of radiation or chemotherapy during pre-transplant conditioning, or from effects of the primary disease or its treatment, including surgery or direct tumor extension. Endocrinopathies are frequently insidious in onset and may not manifest in overt clinical symptoms until reasonably advanced. Failure to recognize (and treat) signs and symptoms of endocrine dysfunction may contribute to impaired quality of life among HSCT survivors; thus, awareness of these potential outcomes is of vital importance for the treating clinician.
Recent estimates suggest that 30 % of patients who have undergone HSCT experience at least one grade 3–5 (classified as “severe, life-threatening or fatal”) endocrine complication, compared to 4.9 % of childhood cancer survivors [2]. In addition, nearly one in every two female survivors of HSCT report ovarian failure (48 %), compared to 4 % of female survivors of childhood cancer treated with conventional chemotherapy. While these data are derived from retrospective cohort studies, reflecting transplant regimens in place between 1974 and 1998, the magnitude of the effect remains sobering.
As with other late effects of HSCT, surveillance and treatment of endocrine complications is adapted to the specific exposures (both pathologic and therapeutic) experienced by the patient. Overall, endocrine manifestations are more common among patients who received TBI as a component of their conditioning regimen, although gonadal toxicity and infertility remain highly prevalent following conditioning using alkylating agents alone (busulfan, cyclophosphamide, melphalan) [5, 95, 96]. Typical doses of TBI (12 Gy for neoplastic disease and 3–8 Gy for non-neoplastic conditions) are unlikely to result in substantial or permanent hypothalamic-pituitary dysfunction. Nevertheless, endocrine late-effects also reflect the combined sequelae of primary disease therapy in combination with treatments administered during the transplant process. Thus, anticipation of these sequelae must take both exposures into consideration.
13.9.1 Abnormalities of Linear Growth
Normal growth results from combined effects of hormonal influences [growth hormone (GH), thyroid hormones and sex steroids], genetic potential and adequate nutrition. Several studies have demonstrated reduced final height among survivors of childhood HSCT [96–98]. Radiation exposure is the primary treatment factor associated with compromise of final adult height. While cranial irradiation in excess of 30 Gy (generally in the context of primary treatment of a CNS malignancy) places patients at increased risk for GH deficiency, TBI also confers an independent risk for growth restriction [99–101]. In contrast to patients who undergo TBI and/or cranial radiation, those exposed exclusively to conditioning with cyclophosphamide or busulfan experience normal growth rates [98, 102–104]. Multiple elements of radiation treatment regimens may compromise final adult height, including the impact of radiation on the hypothalamic-pituitary axis which can result in impairment of GH secretion and pubertal precocity, while peripheral effects include direct radiation toxicity to the vertebral epiphyses [105, 106]. The greatest detrimental effects on growth are experienced by those with prior cranial irradiation and single-dose TBI, while fractionation of TBI helps mitigate the effect [98, 101]. Similarly, age at TBI appears to be a major determinant of height-loss with the most pronounced effect among those receiving radiation prior to age 8 years [107]. Prolonged systemic glucocorticoids may also adversely affect growth.
Due to the complex interplay of factors that guide linear growth, prediction of the likelihood of growth failure is challenging. Nonetheless, the most sensitive diagnostic procedure for assessing early growth failure is serial, precise anthropometry and pubertal staging. Ancillary measures such as bone age measurement and hormonal assessment may be indicated but do not form the basis of screening.
Estimates of GH deficiency in HSCT survivors vary widely from 25 % to 85 % [76, 99, 106, 108]. This wide range reflects differences in conditioning regimens, prior history of cranial irradiation and different methods of GH testing. A substantial interval may exist between timing of HSCT and clinical signs of GH deficiency. For those treated with >30 Gy cranial irradiation [109] (as may be administered for primary CNS malignancy), evidence may be present within 5 years of treatment, while smaller cumulative doses (i.e. 18–24 Gy, given during treatment of CNS relapse for ALL) may result in an interval of ≥10 years [103]. Despite these discrepancies, significant improvement in final height has been observed in children who received GH treatment, irrespective of biochemical evidence of GH deficiency [99, 105, 110]. Children treated with GH prior to 10 years of age have the best response to GH therapy, while male patients are reported to reap less benefit in terms of final height [98, 99]. Additionally, patients with combined GH-deficiency and pubertal precocity (see below) may benefit from pubertal suppression with GnRH agonists to arrest premature epiphyseal closure [111]. Notwithstanding negative effects on growth, several series have demonstrated that a majority of HSCT survivors achieve final heights within 2 standard deviations of the general population, albeit diminished relative to their potential [98, 112, 113].
Given its role in promoting cellular proliferation and mitogenesis, concern has been raised about the risk of tumor recurrence and SMN in cancer survivors treated with GH. Prolonged large-scale follow-up of childhood cancer survivors treated with GH reveals no increased risk of primary tumor recurrence [114–116], although there may be a small but significant increased rate of SMN, primarily meningiomas [117, 118]. While specific data from survivors of HSCT are lacking, given an overall increased incidence of SMN among survivors of HSCT, care should be taken in selecting patients most likely to benefit from GH therapy.
GH deficiency in adults is associated with osteopenia, decreased lean body mass, dyslipidemia, diminished cardiac function as well as decreased quality of life [119]. Thus, replacement therapy may be indicated in adult survivors with a history of GH deficiency, although specific guidelines remain to be established [120–125].
13.9.2 Gonadal Dysfunction and Reproductive Failure
Perhaps no endocrine late-effect of HSCT therapy has as great an impact on quality of life as does gonadal dysfunction and infertility. These effects can manifest at varying stages of patients’ lives including puberty and reproductive years, and stem from damage to both the central (hypothalamic/pituitary) controls and the gonads. Apart from direct physiologic effects on puberty and reproductive health, alterations in gonadal function and fertility may influence body image, interpersonal relationships, sexuality and sense of overall well-being [126, 127]. Moreover, sex steroid deficiency may lead to compromise of other systems such as bone health and linear growth. Although there is clear interplay between effects on fertility and testosterone/estrogen production, consideration of late-effects is facilitated by reviewing these separately.
13.9.3 Puberty and Gonadal Sex Steroid Production
Normal pubertal initiation and progression requires the function of an intact hypothalamic-pituitary-gonadal axis. Resultant stimulation of gonadal sex steroids has effects on osseous maturation, redistribution of body fat, increases in muscle mass, development of secondary sexual characteristics and uterine maturation in girls. While pre-transplant cranial irradiation may result in central (hypogonadotrophic) hypogonadism, far more common among HSCT survivors is primary gonadal failure, resulting from direct gonadal toxicity from radiation and alkylating agent chemotherapy.
Hypogonadism has been reported in approximately a third of long-term survivors of HSCT and is much more common in females (69–95 %) than in males (9–47 %) [128, 129]. In the adolescent, this generally manifests as delayed or dysfunctional puberty, while in adults, hypogonadism may manifest with any of menopausal symptoms in women (menstrual irregularity, hot flashes, vaginal dryness, etc.), diminished libido, altered sexual function, or more vaguely with mood changes or decreased vitality [130].
Normal puberty is considered to be age-appropriate when signs (either breast or testicular enlargement) are first noted between ages 8 and 13 years in girls and between ages 9 and 14 years in boys. Outside of these ranges, pubertal timing may be abnormal and deserving of further evaluation by a pediatric endocrinologist. Absence of menarche by age 16 (or 3 years after first breast development) is also considered abnormal.
Central hypogonadism, due to deficits of LH and FSH secretion, is far less common than GH deficiency among HSCT survivors, but may present following radiation to the sella exceeding 30–40 Gy [131–133]. These doses are most often encountered following primary treatment for a CNS malignancy. Similarly, central pubertal precocity (CPP) is rarely associated with radiation exposures from HSCT alone, but may manifest following cumulative CNS radiation exceeding 18 Gy, presumably by relieving pubertal inhibitory signals within the hypothalamus.
Far more common than central hypogonadism is that resulting from direct gonadal toxicity due to radiation and/or chemotherapy. Treatment regimens incorporating alkylating agents (e.g. busulfan, cyclophosphamide) may compromise testosterone or estrogen production. Males treated with cumulative doses of cyclophosphamide >7.5 g/m2 are at highest risk for testicular toxicity [103]. Testicular radiation doses exceeding 24 Gy are associated with Leydig cell dysfunction and testosterone deficiency [134, 135]. Nevertheless, sex hormone production in boys is more resistant to cytotoxic drugs and radiation than are germ cells and the majority of boys will initiate and complete sexual maturation without hormone replacement. Much smaller exposures are required to compromise male fertility, as will be discussed below.
In contrast, girls are at high risk for ovarian failure following HSCT. There is a 9.3-fold increased risk of ovarian failure as compared to all survivors of childhood cancer and 39.3-fold increased risk compared to the general population [2]. The degree of ovarian compromise is related to age at exposure (with less compromise at younger age at treatment), dose of abdomino-pelvic radiation and chemotherapeutic exposure (specifically, alkylating agents such as procarbazine) [129]. Permanent ovarian failure can result from radiation doses of >6 Gy in adult women, and >10 Gy in girls treated during childhood and adolescence [136]. These exposures are commonly encountered in the course of TBI for neoplastic disease, but may be avoided when transplanting for non-neoplastic conditions. Conventional doses of TBI during conditioning therapy for HSCT result in ovarian dysfunction in almost all girls older than 10 years and half of those younger than 10 years [137].
Female survivors of HSCT who maintain normal ovarian function after treatment are still at risk for premature ovarian failure (early menopause), with dose-dependent risk factors including increasing exposure to pelvic radiation and alkylating agents, as well as older attained age and a diagnosis of Hodgkin lymphoma. Among those exposed to alkylating agents and pelvic radiation, the risk for premature menopause approaches 30 % [136]. Implications of these findings pertaining to family planning should be shared with female survivors, who may then elect to initiate efforts to attain pregnancy (whether naturally, or with assisted reproductive technologies) at a younger age.
There are compelling reasons to support sex hormone replacement for all individuals with testicular or ovarian failure, given their role not only in pubertal induction, progression and sexual health, but also in maintaining adequate growth, cardiovascular fitness, bone health and sense of well-being. Generally, replacement continues until the age of menopause in women and indefinitely in men. Nonetheless, there are limited data on the risks of hormone replacement among cancer-survivors for the extremely long time periods that are indicated in these patients, and there is at least a theoretical possibility that hormone replacement could exacerbate the inherent long-term morbidities associated with childhood cancer treatment including SMN (particularly in hormone-responsive tissues) and coagulopathy. Thus, a careful assessment of risk factors (including family history of BRCA1/2 mutation and Factor V Leiden) is warranted at the outset of therapy. Arguments for and against routine sex hormone replacement in childhood cancer survivors have been reviewed recently [138, 139].
In some instances, gonadal function has been noted to recover after months to years of insufficiency [140]; thus, for individuals receiving hormone replacement, intermittent withdrawal may be indicated to assess for this recovery under the guidance of an endocrinologist or fertility specialist.
13.9.4 Infertility (Germ Cell Failure)
In both sexes, infertility may result from hormone deficiencies as well as direct germ cell damage. The latter can be the consequence of gonadal irradiation (whether TBI, abdominal, pelvic or lumbosacral) and/or alkylating agents used during treatment of the primary disease or in the course of myeloablation prior to transplant. Germ cell compromise is present in the majority of patients treated with cytoreductive chemotherapy or radiation, and infertility is common.
In males, increasing doses of radiation lead progressively to reversible oligo-azoospermia (gonadal doses of 1–3 Gy), azoospermia that is less likely to be reversible (3–6 Gy) or permanent azoospermia (doses >6 Gy) [134, 141]. In a recent study, pre-pubertal therapy was identified as a risk-factor for male infertility, although the magnitude of the effect of age and pubertal status was small [142].
Women who have undergone allogeneic HSCT are unlikely to achieve pregnancy due to the ovarian toxicity of conditioning regimens prior to transplant. Abdomino-pelvic radiation doses as low as 4 Gy are able to destroy up to 50 % of oocytes [143, 144]. Prior cranial irradiation >22 Gy (as may be the case in patients with CNS tumors) also places women at risk for reduced fertility [145]. While there are several reports of pregnancy following HSCT with or without TBI [146–149], those who do become pregnant are at increased risk of miscarriage, premature delivery, and delivery of low- or very low birth weight infants [95, 136, 150]. These complications have not been observed among offspring of male HSCT survivors. There is no increase in rates of malformations or genetic syndromes in offspring of survivors of either gender. The increased miscarriage rate likely results from germ cell compromise as the result of ovarian radiation, while radiation to the uterus has been reported to result in diminished uterine size, muscular elasticity and uterine vascular damage, and this may account for the observed risk of prematurity and low birth weight [151]. In contrast to males, females are at increasing risk for infertility with treatment at advancing age (presumably due to progressive depletion of ovarian reserve) [142].
Measures aimed at fertility preservation in women, including ovarian autotransplant and ovarian wedge resection with cryopreservation and in vitro maturation, are currently being explored, but remain at the investigational stage [152]. Pubertal males maintain the option of sperm banking; however, no such option exists for pre-pubertal boys undergoing treatment. For those boys that are pubertal at the time of an initial oncologic diagnosis, consideration should be given to sperm banking prior to primary therapy if there is a possibility that HSCT may be an eventual treatment modality, even if not first-line.
13.9.5 Thyroid Dysfunction
Thyroid dysfunction following HSCT is common, affecting up to 50 % of survivors [153], compared to 0.6–1.6 % of childhood cancer survivors [154]. Either overt primary hypothyroidism (increased TSH with low free T4) or compensated hypothyroidism (increased TSH with normal free T4) are the most common manifestations. Exposure to single-dose radiation as low as 10 Gy is associated with development of thyroid dysfunction [155, 156]. Cytoreductive chemotherapy without TBI may also be associated with risk of hypothyroidism [157]. Additionally, patients with a history of cGVHD are at 3.2-fold increased risk for development of post-transplant hypothyroidism [158]. In contrast, central hypothyroidism (TSH or TRH deficiency) is rare, unless high doses of cranial irradiation (>40 Gy) are administered [159, 160]. Autoimmune thyroid disease has also been described among HSCT recipients when the donor was affected by thyroid autoimmunity and typically manifests as autoimmune thyroiditis or Graves’ disease [161–163]. Symptoms may appear several months to years after transplant with no obvious plateau in risk, even 25 years after treatment [154]. Furthermore there is a significant increase in the appearance of thyroid nodules and primary thyroid malignancy among survivors treated with radiation doses up to 30 Gy [164]. Survivors treated with radiation to a field that involves the thyroid (including TBI) require annual assessment of their thyroid function as well as examination of the thyroid.
13.9.6 Bone Health and Complications Among HSCT Survivors
Multiple components of primary disease therapy, as well as that involved in HSCT place patients at risk for compromised bone health. These include radiation therapy, prolonged systemic glucocorticoids and other chemotherapeutics (e.g. methotrexate) and, when superimposed, deficiencies in growth hormone and sex steroids. As a result, survivors of childhood cancer therapy and of HSCT are at increased risk for decreased bone density and osteonecrosis.
13.9.7 Diminished Bone Mineral Density (BMD)
Multiple studies have demonstrated a reduction in overall bone mineral density (BMD) among survivors of HSCT, particularly that population treated for cGVHD [165–167], although this finding is not replicated in all cohorts [168, 169]. Predictors of low BMD include dose and duration of glucocorticoid treatment, duration of treatment with Cyclosporine A or tacrolimus, older age at transplant and female gender [166, 170]. Race, primary diagnosis and time from transplant do not appear to be associated with risk. Although there is some recovery of BMD following treatment, survivors remain at increased risk for low BMD through adulthood [171, 172]. There is some suggestion that treatment with bisphosphonates prior to [173] or following [174–176] conditioning may preserve bone density, although studies are small and have not been performed among children undergoing HSCT. Accepted preventative therapeutic modalities include optimization of Calcium and Vitamin D intake, encouragement of weight-bearing exercise and identification (and treatment) of underlying growth hormone and sex steroid deficiencies. BMD assessment by DEXA or quantitative CT is recommended for all patients at entry to long-term follow-up and periodically thereafter [171]. Results of DEXA analysis in children must be interpreted according to age, pubertal status and height, using Z-scores rather than T-scores (which are the standard among adults). Failure to use appropriate comparators may result in overdiagnosis of low BMD [169, 177]. Moreover, in contrast to the adult literature, there is little pediatric evidence to support extrapolation of BMD to future fracture risk.
13.9.8 Osteonecrosis
Prolonged glucocorticoid therapy, most notably in the setting of cGVHD, places patients at increased risk for osteonecrosis (avascular necrosis). Osteonecrosis (ON) results from diminished blood supply and bone marrow ischemia. Symptoms include pain, swelling, immobility and decreased range of motion. In one study, osteonecrosis was noted on MRI in 44 % of children who had undergone HSCT [170]. Other factors associated with increased risk of osteonecrosis include TBI and age >10 years at the time of transplant [178–181]. In one study, children who underwent HSCT who did not develop cGVHD were no more likely than their siblings to develop osteonecrosis, while those with a history of GVHD developed ON at significantly higher rates [158]. Children carrying a polymorphism of the PAI-1 gene have been found to be at increased risk for ON following HSCT [182]. While the hip is the most commonly affected joint, other commonly affected joints include the humerus and other weight bearing long-bones. Annual screening via musculoskeletal examination is indicated, and suspicion of ON should prompt evaluation with MRI. MRI, however, has not yet been demonstrated to be an appropriate modality for routine screening. Apart from limiting glucocorticoid exposure, wherever possible, there is little to help prevent development of ON. Treatment requires consultation with an orthopaedic surgeon and may include physical therapy, analgesia and/or surgical intervention. There is recent evidence from limited non-controlled studies supporting the use of bisphosphonate therapy for symptomatic treatment of ON [183, 184].
13.9.9 Metabolic Consequences of HSCT
13.9.9.1 Overweight and Obesity
Survivors of acute lymphoblastic leukemia (ALL) are at increased risk for becoming overweight or obese [185, 186], though there is little data to suggest any additional risk accrued during the course of HSCT. Although one study demonstrated increased fat mass in survivors of HSCT when compared to controls (34.9 % fat mass vs. 24.3 %) [187], all patients in this study carried primary diagnoses of ALL or lymphoblastic lymphoma. Moreover, another study demonstrated a propensity towards underweight in HSCT survivors [188]. Mechanisms of altered body composition in these populations may include GH deficiency and disruption of hypothalamic satiety and energy-regulating centers. Additionally, restrictions of physical activity due to hospitalization and illness during the periods of primary disease treatment and HSCT may contribute to altered BMI.
13.9.9.2 Metabolic Syndrome
Survivors of HSCT are at increased risk for developing the metabolic syndrome (MS), defined by the National Cholesterol Education Program as any three of central obesity, hypertriglyceridemia, low HDL-cholesterol, elevated blood pressure, or increased fasting glucose. A recent study estimated the relative risk for MS among HSCT survivors at 2.2-fold over the general population [189–191]. Among survivors of childhood leukemia, HSCT with TBI (and not HSCT without TBI) was the only factor demonstrated to have a significant association with development of the MS [192]. Dyslipidemia may develop in up to 28 % of survivors [193]. Further biochemical comparison of this population with those in the general population with MS highlights significantly higher levels of leptin, CRP and TNF-α [194], suggesting possible underlying hypothalamic (e.g. hyperleptinemia) and/or systemic inflammatory mechanisms. Other proposed mechanisms for development of MS include endothelial dysfunction and nutritional insults during vulnerable developmental stages, both of which are plausible mechanisms in the face of treatment and disease-related stresses of HSCT [185]. While increased risk for the MS may partially explain the increased cardiovascular complications reported among survivors of cancer [195, 196] and HSCT [2], improvement in the parameters of MS has not yet been demonstrated to decrease CV risks in this population.
13.9.9.3 Diabetes
Although perturbations in glycemic control are common in the immediate post-transplant period, long-term effects on glycemia are less-well characterized [197–199]. Survivors of allogeneic HSCT were 3.7 times more likely to report diabetes than siblings [200], while childhood cancer survivors treated with conventional therapy were 1.8 times more likely to report diabetes than siblings [201]. In a recent pilot study, 5 out of 10 young adult survivors of HSCT had biochemical evidence of diabetes despite the absence of clinical symptoms, suggesting a high rate of occult disease [202]. Similarly, hyperinsulinemia without hyperglycemia (i.e. a state of insulin resistance) was noted following an intravenous glucose tolerance testing in a cohort of 24 survivors of childhood HSCT who underwent TBI, but not those who had isolated thoraco-abdominal radiation or who were not radiated, suggesting that the effect was generalized and not solely a result of pancreatic damage [203].
Diabetes among HSCT survivors appears to be distinct from both Type I and Type II diabetes. It is predominantly characterized by insulin resistance, rather than diminished beta-cell function, although patient characteristics are distinct from those classically associated with Type II diabetes [197, 198, 200, 202–204]. Specifically, among HSCT survivors that develop diabetes, BMI is not necessarily elevated, there is rarely a positive family history of diabetes and patients do not necessarily derive from “high-risk” ethnic groups. Diabetes in HSCT survivors is invariably insidious in onset and rarely associated with diabetic ketoacidosis [205]. Development of post-transplant diabetes was reported in a recent cohort in 28/369 (7.6 %) patients, although this estimate was based on retrospective analysis and may represent an under-diagnosis, given the nature of the disease [128]. As with the metabolic syndrome, post-transplant diabetes has not been specifically demonstrated to accrue the same risk of micro- and macrovascular disease as “conventional” Type I or Type II diabetes.
13.10 Cardiac Late Effects
Therapy with anthracyclines and treatment with radiation to a field that involves the heart can lead to cardiac disease in childhood cancer survivors. Many children who undergo HSCT will be exposed to these cardiotoxic therapies prior to transplant. Their risk for cardiac disease can be compounded by their transplant conditioning (e.g. TBI) [206]. Although high-dose cyclophosphamide has been proposed as a possible risk factor for cardiac toxicity, the evidence for this is limited [207]. Anthracycline agents are most commonly linked to myocardial damage with an increased risk for reduced cardiac function that, in severe cases, can manifest as congestive heart failure. Radiation therapy can also damage the myocardium, and it is also associated with coronary artery disease, pericarditis, valvular disease, and conduction abnormalities. As discussed above, HSCT survivors are also at risk for metabolic sequelae of their transplant such as diabetes mellitus, hypertension [207] and lipid abnormalities [192] that can impact negatively on their cardiac health. One study demonstrated that the presence of two or more of obesity, dyslipidemia, hypertension or diabetes in survivors of adult HSCT was associated with a five-fold increased risk of cardiovascular disease [208].
Survivors who have been treated with an anthracycline or chest radiation (including TBI) require cardiac imaging (by echocardiogram or MUGA) every 1–5 years, depending on their age at treatment, cumulative anthracycline dose, and radiation exposure [209]. Care providers should be aware of their risks for metabolic sequelae and should assess weight and blood pressure at all visits, and screen for glucose intolerance or dyslipidemia periodically.
Related posts:
 Endocrine System
Endocrine System
 Long-Term Follow-Up Roadmaps by Disease and Treatment
Cultural and Linguistic Issues in the Assessment and Treatment of Pediatric Cancer Survivors
Career and Vocational Outlook
Long-Term Follow-Up Roadmaps by Disease and Treatment
Cultural and Linguistic Issues in the Assessment and Treatment of Pediatric Cancer Survivors
Career and Vocational Outlook
 Neuropsychological Effects of Pediatric Brain Tumors and Associated Treatment
Neuropsychological Effects of Pediatric Brain Tumors and Associated Treatment
 Educating and Preparing the Childhood Cancer Survivor for Long-Term Care: A Curriculum Model for Cancer Centers
Educating and Preparing the Childhood Cancer Survivor for Long-Term Care: A Curriculum Model for Cancer Centers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


