Feature
Type of heavy-chain disease
α
γ
μ
Year described
1968
1964
1969
Incidence
Rare
Very rare
Very rare
Age at diagnosis
Young adult (<30 years)
Older adult (60–70 years)
Older adult (50–60 years)
Geographic region
Mediterranean
Worldwide
Worldwide
Structurally abnormal monoclonal protein
IgA
IgG
IgM
MGUS phase
No
Rarely
Rarely
Urine monoclonal light chain
No
No
Yes
Urine abnormal heavy chain
Small amounts
Often present
Infrequent
Sites involved
Small intestine, mesenteric lymph nodes
Lymph nodes, bone marrow, spleen
Lymph nodes, marrow, liver, spleen
Pathology
Extranodal marginal-zone lymphoma (MALT or IPSID)
Lymphoplasmacytoid lymphoma
Small-lymphocytic lymphoma, CLL
Associated diseases
Infection, malabsorption
Autoimmune diseases
None
γ-Heavy-Chain Disease
Since the first report of γ-HCD in 1964 [2], approximately 130 patients with this disease have been described in the medical literature [3–5]. A lymphoma-like illness, with lymphadenopathy, splenomegaly, and hepatomegaly together with a lymphoplasmacytic proliferation similar to that of Waldenström macroglobulinemia, has been considered characteristic of γ-HCD. However, it does not represent a specific pathologic process; it is a biochemical expression of a mutant B-cell clone. The “disease” should be considered a serologically determined entity.
Epidemiologic Factors
γ-HCD has been described throughout the world, and no epidemiologic pattern has been recognized. No familial cases have been reported. Although initially γ-HCD was reported to occur equally in men and women [3], a clear predominance of women was found in a more recently described series of 23 patients [5]. The median age at diagnosis in that series was 68 years (range, 42–87 years).
Pathogenesis
The pathogenesis of γ-HCD is unknown.
Clinical Features
γ-HCD disease can present with a great variety of clinical and pathologic features. Integrating these various changes into a unified disease concept has been difficult. Wester et al. [6] suggested that patients with γ-HCD can be placed into three broad categories on the basis of the underlying pathologic process and its distribution (1) those with disseminated lymphoproliferative disease (approximately 66 % of patients); (2) those with localized lymphoproliferative disease (approximately 25 %; 11 % with extramedullary involvement and 14 % with bone marrow involvement only); and (3) those with no apparent proliferative disease (approximately 9 %). Of the patients with no proliferative disease, many have autoimmune disorders [6].
Disseminated lymphoproliferative disease is present in most patients at diagnosis and has been reported in 57–66 % of patients in various series [3, 5, 6]. Lymphadenopathy and constitutional symptoms, such as fever, weakness, and fatigue, are the most common initial features. On physical examination at diagnosis in two different series [3, 5], lymphadenopathy was present in 56 % and 62 % of patients, splenomegaly in 38 % and 52 %, and hepatomegaly in 8 % and 37 %. Lymphadenopathy obstructing the vena cava has been noted [7, 8]. Waxing and waning of lymphadenopathy may occur. Edema of the uvula and palate occurs in about 20 % of patients, presumably because of lymphatic obstruction due to involvement of the lymph nodes of the Waldeyer tonsillar ring. When present, splenomegaly ranges from a palpable tip to massive splenomegaly. Isolated splenomegaly may be the presenting feature of γ-HCD [3]. Spontaneous rupture of the spleen has been described [3].
Several extrahematopoietic organs have had tumors in γ-HCD. Skin lesions in general have been noted to be the most frequent extrahematopoietic manifestation [5, 9]. Cutaneous or subcutaneous involvement, featuring an extranodal mass [3, 10, 11] or skin nodules [8–10, 12–18], has been described. Patients have presented with extramedullary plasmacytoma of the thyroid [5, 10, 19–22], parotid or submandibular swelling [2, 10, 23], or an oral pharyngeal mass [5, 24]. A case with hypertrophic spinal pachymeningitis has been reported [25]. A 12-year-old Turkish girl presented with malabsorption and marked infiltration of the intestine with lymphoplasmacytoid cells (similar to α-HCD) [26], and a 63-year-old woman who had γ-HCD with mucosa-associated lymphoid tissue lymphoma of the duodenum has been reported in the Japanese literature [27]. In several other cases, the initial symptoms were due to a gastric lymphoid tumor [28–30].
The occurrence of autoimmune disorders with or without associated underlying lymphoid proliferation is frequent in patients with γ-HCD. Rheumatoid arthritis has been reported in about a dozen patients [3, 5, 14, 31–42] and preceded the diagnosis of γ-HCD by as many as 25 years. Two patients with rheumatoid arthritis had only transient monoclonal γ-heavy chains [33, 40]. Systemic lupus erythematosus was documented in one case [43] and discoid lupus in another [19]. Vasculitis with or without associated rheumatoid arthritis [3, 35, 39] and livedoid vasculitis [44] have been reported. Sjögren syndrome was present in three cases [5, 45, 46], and keratoconjunctivitis sicca, vasculitis, hypocomplementemia, and Coombs-positive hemolytic anemia were noted in another [47]. Two patients with myasthenia gravis have been described [3, 19]. Peripheral neuropathy has been reported in several patients [3, 48, 49] and progressive multifocal leukoencephalopathy in one patient [50], and another patient had a past history of Guillain-Barré syndrome [10].
Among solid tumors that have been diagnosed in patients with γ-HCD are prostate cancer [10, 48], uterine sarcoma [3, 32], gastric carcinoma [3], and pancreatic cancer [51]. These tumors are probably fortuitous. A myeloproliferative disorder was documented in three cases [6, 52, 53]. Herpes zoster involved the eye of one patient [54] and was generalized in another [55].
Laboratory Findings
Hematologic Abnormalities
Anemia is frequent and ultimately develops in nearly all patients with γ-HCD. It usually is normochromic, normocytic, and moderate except in patients with autoimmune hemolytic anemia. A Coombs-positive autoimmune hemolytic anemia has been reported in several cases [5, 10, 12, 36, 47, 56, 57] and has sometimes been associated with idiopathic thrombocytopenic purpura (Evans syndrome) [5, 10, 12]. The leukocyte count and differential cell count are usually normal, but both leukopenia and leukocytosis may occur. Lymphocytosis with or without atypical lymphocytes may be present, and occasionally a patient presents with chronic lymphocytic leukemia. Plasmacytoid lymphocytes or plasma cells may be found in the peripheral blood, and plasma cell leukemia has been reported in two patients [58, 59]. Eosinophilia may occur. Thrombocytopenia due to an autoimmune process, hypersplenism, and, less frequently, bone marrow failure has been reported, but thrombocytosis is not a feature of γ-HCD. The erythrocyte sedimentation rate may vary from within the reference range to more than 100 mm in 1 h.
Bone Marrow Findings
Bone marrow aspirates and biopsy specimens frequently show an increase of plasma cells, lymphocytes, or plasmacytoid lymphocytes, which is similar to the bone marrow picture of Waldenström macroglobulinemia. Typical bone marrow features of multiple myeloma [5, 60, 61] or chronic lymphocytic leukemia [3, 5, 10] are rare. Marrow changes consistent with a myeloproliferative disorder have been noted in a few patients [5, 6, 52, 53]. Eosinophilia is rarely found. The presence of mast cells has been mentioned [62]. A marked increase in erythropoiesis is present in patients with hemolytic anemia. In several instances, the bone marrow aspirate and biopsy specimens have shown normal results.
Other Features
Bone lesions are very rare in γ-HCD; only four cases with skeletal involvement have been reported [3, 15, 16, 61, 63]. Hypercalcemia was noted in five patients [7, 10, 16, 17, 64], three of whom had no apparent skeletal involvement. Renal insufficiency is uncommon but may occur in association with hypercalcemia. Lymphoid infiltration involving the kidneys [10, 57], adrenal glands [11, 65], lungs [10, 18], and the central nervous system [35] has been detected, mainly at postmortem examinations.
Protein Findings
The serum protein electrophoretic pattern is extremely variable. A monoclonal peak is detected in 60–86 % of γ-HCD patients [3, 5]. In approximately 15–40 % of patients, the abnormal protein is not detectable by electrophoresis [3, 5], and the pattern is normal, shows hypogammaglobulinemia, or shows hypergammaglobulinemia with a polyclonal pattern or an increase in the protein migrating in the β region without a detectable spike. The range of mobility of the γ-HCD protein is wide; an abnormal band in the β region is the most common pattern. The serum level of the γ-HCD protein is usually less than 1 g/dL but can range from an immeasurable level to 9 g/dL [3]. The median value of the monoclonal spike at diagnosis in 19 patients was 1.59 g/dL (range, 0.40–3.91 g/dL) [5].
No gold standard has been established to identify the subclass of the heavy-chain fragment. In reported cases in which a heavy-chain fragment subclass has been studied, different methods have been used, ranging from Ouchterlony technique in the earlier cases to indirect immunofluorescence staining, immunoblotting, amino acid sequencing, immunoselection, and enzyme-linked immunosorbent assay [66]. Analysis of the distribution of the immunoglobulin G (IgG) subclasses in γ-HCD shows a lower-than-expected incidence of IgG2 HCD protein. The most common subclass is IgG1, which occurs in 65 % of cases; IgG3 has been identified in 27 %, IgG4 in 5 %, and IgG2 in 3 % [3]. By comparison, the normal distribution of IgG subclasses is IgG1, 64–70 %; IgG2, 23–28 %; IgG3, 4–7 %; and IgG4, 3–4 % [67]. Immunoglobulin A (IgA) and immunoglobulin M (IgM) are usually reduced in γ-HCD.
The response to the Bence Jones heat test is in general negative. Occasionally, a patient with γ-HCD and Bence Jones proteinuria has been described [5, 36, 68]. The amount of γ-HCD protein excreted in the urine is less than 1 g in 24 h in most instances but may reach 20 g in 24 h. In many cases, careful study of adequately concentrated urine is necessary to detect the γ-HCD protein. The mobility of the monoclonal protein in urine is the same as that in serum, with rare exceptions only [7].
Relatively homogeneous free fragments of γ chains unaccompanied by light chains may be found during examination of urine through high-resolution electrophoresis and immunofixation. This urinary protein, which should not be confused with the γ-HCD protein [69], is similar to the fragment generated by the secondary action of papain on the Fc fragment. Differentiation from the γ-HCD protein can be accomplished by gel diffusion analysis. From a practical point of view, however, such studies are not required because the γ-HCD protein is always present in the serum and migrates in the β–γ region, whereas the free γ-chain fragments described by Charles and Valdes [69] are found only in the urine and migrate in the α2 region. These fragments are of no known clinical significance. They probably originate from extracellular degradation of the IgG molecule.
Cytogenetic Abnormalities
Cytogenetic abnormalities have been recognized infrequently. They have been reported in 7 of 15 patients, of whom four had aneuploidy [3, 10, 13, 32] and the other three had trisomy 7 [18], trisomy 21 [70], and multiple chromosome abnormalities [3]. Three of these seven patients [3, 32] had received prior chemotherapy, which could have been responsible for some of the chromosome abnormalities. No chromosome abnormalities were found in seven other patients [2, 3, 23, 31, 33, 71].
Structural Protein Abnormalities
HCD proteins are synthesized as truncated chains. In some cases, an additional, limited postsynthetic degradation occurs with removal of the N-terminal sequences, which can be documented in the proliferating cells. Immunochemical studies indicate that the HCD proteins have intact carboxyl-terminal regions. The alterations most often involve the V and the CH1 domains. The precise boundaries of the deletions vary from one case to another. Structural studies have shown that most γ-HCD proteins are dimers of deleted heavy chains devoid of light chains and consist mainly of the Fc region. The molecular weight of the monomeric γ chain ranges from 27,000 to 49,000. The carbohydrate content of the γ-HCD proteins is high. The length of the γ chain varies from case to case but usually is one-half to three-fourths the length of its normal counterpart [72]. Often, the deletions are internal, with a portion of the V sequence present at the amino terminus, ruling out postsynthetic degradation. The resumption of the normal sequence occurs precisely at the beginning of a domain. On the basis of their structure, the γ-HCD proteins can be placed into several groups (Fig. 36.1).
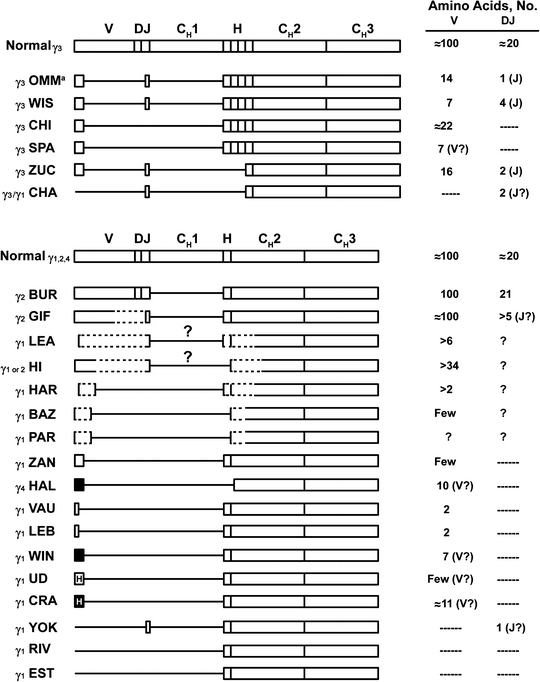
Fig. 36.1
Structure of various deleted γ-heavy-chain disease (HCD) proteins compared with normal chains.  indicates heterogeneous amino acid sequences;, unusual amino acid sequences;
indicates heterogeneous amino acid sequences;, unusual amino acid sequences;  , unusual and heterogeneous amino acid sequences;
, unusual and heterogeneous amino acid sequences;  boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; dashed lines, likely structures for which sequence data are missing; H hinge region; J joining region; lines, deletions; question mark, probable missing domain based on molecular weight and partial protein structure analysis; V variable region. aStructure shown is a primary synthetic product synthesized by the HCD cells. Serum protein was modified after synthesis and did not contain any amino acids before the hinge. Citations for proteins are OMM [51, 73], WIS [74, 75], CHI [76], SPA [77], ZUC [78], CHA [79], BUR [80], GIF [81], LEA [82], HI [83], HAR [82], BAZ [84], PAR [85], ZAN [75], HAL [86], VAU and LEB [87], WIN [68], UD [88], CRA [82, 89], YOK [90], RIV [91], and EST [92] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)
boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; dashed lines, likely structures for which sequence data are missing; H hinge region; J joining region; lines, deletions; question mark, probable missing domain based on molecular weight and partial protein structure analysis; V variable region. aStructure shown is a primary synthetic product synthesized by the HCD cells. Serum protein was modified after synthesis and did not contain any amino acids before the hinge. Citations for proteins are OMM [51, 73], WIS [74, 75], CHI [76], SPA [77], ZUC [78], CHA [79], BUR [80], GIF [81], LEA [82], HI [83], HAR [82], BAZ [84], PAR [85], ZAN [75], HAL [86], VAU and LEB [87], WIN [68], UD [88], CRA [82, 89], YOK [90], RIV [91], and EST [92] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)
indicates heterogeneous amino acid sequences;, unusual amino acid sequences; , unusual and heterogeneous amino acid sequences; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; dashed lines, likely structures for which sequence data are missing; H hinge region; J joining region; lines, deletions; question mark, probable missing domain based on molecular weight and partial protein structure analysis; V variable region. aStructure shown is a primary synthetic product synthesized by the HCD cells. Serum protein was modified after synthesis and did not contain any amino acids before the hinge. Citations for proteins are OMM [51, 73], WIS [74, 75], CHI [76], SPA [77], ZUC [78], CHA [79], BUR [80], GIF [81], LEA [82], HI [83], HAR [82], BAZ [84], PAR [85], ZAN [75], HAL [86], VAU and LEB [87], WIN [68], UD [88], CRA [82, 89], YOK [90], RIV [91], and EST [92] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)Most γ-HCD proteins contain a fragment of a V region. Usually, they have a normal V region amino terminus followed by an internal deletion of the V and the entire CH1 domain. In all HCD proteins with an internal V-region deletion, the residues corresponding to the VDJ junction are missing. Some of these proteins contain a portion of this region and are featured by two noncontiguous deletions (proteins γ3 OMM, γ3 WIS, and γ3 ZUC). The γ3 OMM protein has been shown to undergo postsynthetic degradation to yield an NH2-terminal deleted protein [93]. Some γ-HCD proteins lack the entire V and CH1 domains, with the sequence starting with the hinge region (γ1 RIV, γl EST).
The third group is characterized by an unusual amino acid sequence preceding the deletion. An abnormal sequence of ten residues has been found in protein γl CRA. The 7 N-terminal amino acid residues found in protein γl WIN are not translated from any of the known immunoglobulin heavy-chain gene sequences [68]. The complete sequence of γ2-HCD BUR has been described [80]. This mutant is composed of a complete V region, hinge, CH2, and CH3 domains. The unique features of this V region are the presence of methionine at position 11, two cysteine residues at positions 50 and 53, and three glycosylation sites.
In most cases of HCD, the association of several distinct gene alterations is responsible for both the complex structural abnormalities found among these proteins and the usual absence of light-chain synthesis. The alterations found in two γ-heavy-chain genes (γ-HCD protein OMM and γ-HCD protein RIV) and in one light-chain gene (κ RIV) include somatic mutations, deletions, and insertions in rearranged V genes. The HCD protein γ3 OMM was found to be the product of two deletions—a splice correction and postsynthetic NH2-terminal proteolysis [73]. Sequencing of the γl RIV gene showed that it had undergone VH–JH and H chain-class switch recombinations. However, normal RNA splice sites had been eliminated by a DNA insertion–deletion (VH acceptor site), mutations (JH donor site), or a large deletion (CH1 region). These DNA alterations resulted in aberrant mRNA processing, in which the leader region was spliced directly to the hinge region, accounting for the HCD protein [91] (Fig. 36.2).
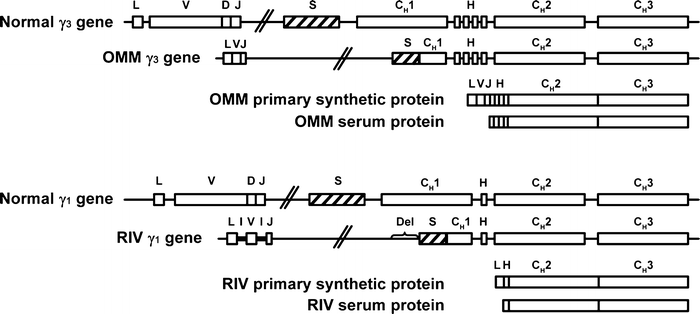
Fig. 36.2
Structure of two genes coding for γ-heavy-chain disease (γ-HCD) proteins compared with normal γ3 and γ1 genes.  indicates switch region;,
indicates switch region;,  inserted noncoding sequence; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; Del deleted sequence; H hinge region; I inserted sequence; J joining region; L leader region; lines, intervening (noncoding) sequences; S switch region; V variable region. Citations for proteins are OMM [51, 73] and RIV [91] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)
inserted noncoding sequence; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; Del deleted sequence; H hinge region; I inserted sequence; J joining region; L leader region; lines, intervening (noncoding) sequences; S switch region; V variable region. Citations for proteins are OMM [51, 73] and RIV [91] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)
indicates switch region;, inserted noncoding sequence; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; Del deleted sequence; H hinge region; I inserted sequence; J joining region; L leader region; lines, intervening (noncoding) sequences; S switch region; V variable region. Citations for proteins are OMM [51, 73] and RIV [91] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)Similar to genes encoding HCD proteins, the κ-chain gene of patient RIV showed deletions, insertions, and a high rate (25 %) of somatic mutations in the VJ region. Mutations of spliced sites bounding the VκJκ region resulted in exon skipping and splicing of the leader peptide exon onto the Cκ exon. A short mRNA was present and coded for a Cκ fragment devoid of the V domain [94].
Usually, γ-HCD cells do not secrete light chains. However, a few cases have been reported in which light chains or light-chain fragments were detected in serum and urine through immunofluorescence or internal labeling. In γ-HCD patient WIN, λ Bence Jones protein was found in serum and urine [68, 95], and IgGκ and IgMλ and κ and λ Bence Jones proteins were detected in another patient’s serum and urine, respectively [36]. In a patient described by Richter et al. [96], the plasma cell proliferation produced truncated γ3 heavy chains and a subpopulation of cells (30 %) secreted large quantities of free κ light chains. In one (patient MO) of nine patients with γ-HCD, nonsecreted monotypic light chains were found through direct immunofluorescence in the cytoplasm and on the surface of blood and bone marrow cells [97]. Small amounts of a truncated κ chain corresponding to the C domain were found in cytoplasmic extract through internal labeling when studying HCD protein RIV. This light-chain fragment was undetectable with immunofluorescence [94, 98].
Heavy- and light-chain assembly is critical for immunoglobulin secretion, and unless deleted, free heavy chains are not secreted in the absence of light chains. Under these circumstances, entire heavy chains continue to be associated with a heavy-chain binding protein in the lumen of the endoplasmic reticulum and are not secreted [99]. However, truncated heavy chains appear to escape the interaction with binding protein and are secreted or expressed at the cell surface in the absence of light chains. Thus, any mutation or deletion impeding the normal folding, assembly, or intracellular transport of immunoglobulin chains could select for the various gene alterations seen in HCD, to maintain immunoglobulin secretion [98, 100]. Although these observations favor a concept in which the light-chain abnormality occurs first, the complexity of the genomic abnormalities described makes a multitude of alterations occurring in the most fragile regions of the immunoglobulin genes more likely.
Oncogene activation has been shown in one patient with γ-HCD (patient OMM) [101]. DNA from a lymphoid line generated from peripheral blood of the patient transformed NIH/3 T3 cells, which produced tumors in white mice. Molecular studies showed an activating N-Ras 61 mutation, as has been noted in one-third of patients with multiple myeloma.
Lymph Node Pathology
In contrast to α-HCD, γ-HCD has no consistent morphologic pattern. The most frequent histopathologic finding is a pleomorphic, malignant lymphoplasmacytic proliferation in marrow and lymph nodes. These lymphocytoid plasma cells express pan-B-cell markers and cytoplasmic γ-heavy chains without light chains and are negative for CD5 and CD10 [1].
Non-Hodgkin lymphoma without a consistent morphologic type was diagnosed in 18 (38 %) of 47 patients for whom lymph nodes were examined [6]. Lymphoplasmacytic proliferation, with or without atypia, was present in 36 % of cases. Hyperplastic nodes and plasmacytoma each made up 11 % of the total. There was one case of Hodgkin disease and one case of “probable” Hodgkin disease [6]. Additional cases of γ-HCD associated with Hodgkin disease have been reported [5, 13, 102–104]. In two cases, the subtype was nodular sclerosing [13, 102]; in one case, mixed cellularity [102]; in one case, nodular lymphocytic predominance [104]; and in one case, lymphocytic depletion [5].
Diagnosis
γ-HCD can present with diverse clinical features. The diagnosis can be overlooked easily on serum protein electrophoresis because a narrow abnormal band suggestive of a monoclonal protein is seen in less than 50 % of patients. Proteinuria may not be detectable. The diagnosis is established through immunofixation or immunoelectrophoresis (Fig. 36.3) of the serum and of a concentrated urine specimen with the use of specific antisera. A modified immunoselection technique for the diagnosis of HCD has been described [105]. Two-dimensional electrophoresis and immunoblotting also have been used for the recognition of γ-HCD [106, 107]. Tissot et al. [108] showed that the combination of serum protein agarose electrophoresis and two-dimensional electrophoresis can be used to further characterize abnormal protein bands detected by immunofixation. In one case of γ-HCD, low concentrations of free heavy chains in serum were detected with capillary zone electrophoresis coupled with immunosubtraction [109].
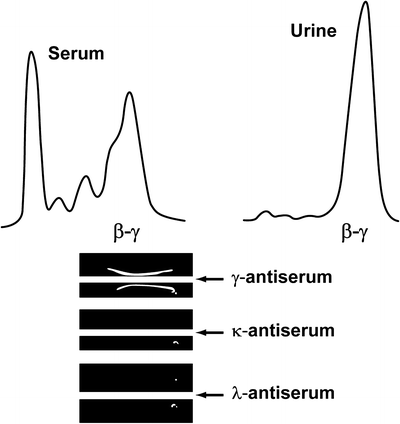
Fig. 36.3
Densitometry tracing and immunoelectrophoresis of a patient with γ-heavy-chain disease. Top, Densitometer tracings with peaks of β–γ mobility in serum and urine profiles. Bottom, Immunoelectrophoresis. In each tray, the top well contained serum and the bottom well contained concentrated urine. With γ antiserum, both serum and urine formed dense, thick, asymmetrical arcs; with κ and with λ antisera, serum and urine formed no visible arcs. This patient had monoclonal γ chain in serum and urine (γ-heavy-chain disease) (Adapted from Kyle RA, Greipp PR. 3. The laboratory investigation of monoclonal gammopathies. Mayo Clin Proc. 1978;53:719–739. Used with permission of Mayo Foundation for Medical Education and Research.)
We believe that γ-HCD is underdiagnosed and urge that immunofixation of serum and urine be performed in all cases of lymphoplasma cell proliferative diseases.
Treatment
Because γ-HCD is a heterogeneous condition that does not really deserve the designation of a disease process and, as suggested by Roda et al. [13], should rather be named “monoclonal γ-HCD protein associated with…,” the choice of therapy should rely entirely on the underlying disorder and the pathologic findings. For an asymptomatic patient with a monoclonal gammopathy of undetermined significance, no therapy is indicated. Any associated autoimmune disease should be managed with standard therapy without taking into account the existence of the abnormal monoclonal component. In symptomatic patients with a low-grade lymphoplasmacytic malignancy, a trial of chlorambucil may be beneficial. Melphalan and prednisone can be used when the proliferation is predominantly plasmacytic. We recommend a trial of cyclophosphamide, vincristine, and prednisone for patients with γ-HCD and evidence of a progressive lymphoplasma cell proliferative process or high-grade non-Hodgkin lymphoma. If no response is obtained with this regimen, doxorubicin should be added.
Agrawal et al. [110] described a patient who achieved complete remission with disappearance of the heavy chain after six courses of fludarabine. Successful treatment of γ-HCD with low-dose etoposide has been reported in a Japanese patient whose disease was unresponsive to combination chemotherapy [111]. Analysis of CD20 expression has been reported in only seven cases and was detected in six of the seven, including one patient in whom CD20 expression appeared transient [5, 112–114]. Rituximab monotherapy was given in two cases, resulting in clinical response [5, 112]. In another patient, the combination of rituximab with chemotherapy (cyclophosphamide, etoposide, procarbazine, and prednisone) showed abundant antitumor effect in lymphoplasmacytic-type γ-HCD [113]. There are no reports of patients treated with thalidomide, bortezomib (Velcade), lenalidomide (Revlimid), or autologous stem-cell transplantation.
The amount of serum γ-HCD protein usually parallels the severity of the associated malignant process. Disappearance of the monoclonal component from serum and urine has been induced with chemotherapy [3, 110], radiation therapy [3, 22], or surgical removal of a localized process [20, 21]. In a few instances, however, relapse is not accompanied by reappearance of the pathologic protein [3]. In general, therapeutic responses have been variable and disappointing because responses often are incomplete or of short duration.
Clinical Course
As expected from the diverse disease picture, the course of γ-HCD varies from an asymptomatic presence of a monoclonal heavy chain in serum or urine to a rapidly progressive downhill course. The presence of a γ-HCD protein alone does not influence the prognosis. Patients with features of a benign monoclonal gammopathy or monoclonal gammopathy of undetermined significance have continued to be clinically well during 2–7 years of follow-up evaluation [5, 19, 115–117].
Disappearance of the γ-HCD protein also has been reported [3, 22, 33, 62, 118]. In one patient, the protein was detected during a recurrent febrile illness but disappeared after a cholecystectomy [62, 118]. In another patient, in whom biopsies had shown an invasive plasmacytic proliferation, the monoclonal γ-heavy chain disappeared from the serum 8 months after irradiation for a thyroid mass and cervical lymphadenopathy [22]. In two patients with rheumatoid arthritis, the γ-HCD protein disappeared [3, 33]. One of these patients has been observed for more than 10 years, and despite slowly progressing rheumatoid arthritis, the γ-HCD protein has not reappeared and an overt lymphoproliferative disorder has not developed [3].
When an underlying lymphoid malignancy exists, the histopathologic pattern is essential for the determination of prognosis. Clearly, patients with an aggressive malignant process do less well than those with more favorable pathologic findings.
Monoclonal Immunoglobulin and γ-HCD
Biclonal gammopathy is reported to occur in approximately 1–3 % of all patients with serum M components [119–121]. In contrast, the association of γ-HCD and another monoclonal gammopathy is much greater. In a review of 56 cases of γ-HCD, nine cases (16 %) of biclonality were identified [10]. In a series of 23 patients with γ-HCD, 7 % were found to have IgMλ intact monoclonal immunoglobulin [5]. In a review by Presti et al. [122], the average age of 13 patients with biclonal γ-HCD was 56 years, and male patients predominated 2:1 [122]. The associated monoclonal immunoglobulin has been of the IgM [3, 5, 29, 30, 34, 59, 122–125] or IgG type [35, 51, 126–130]. No association between γ-HCD and a monoclonal IgA has yet been described, whereas the IgG–IgA association was the most frequent in series of biclonal gammopathies [119, 131]. The frequency of λ chain expression was greater than κ chain expression [122]. All cases except for one case [35] were associated with a lymphoplasma cell proliferative process. The median duration of survival for patients with biclonal disease was 22 months [122]. One patient described by Lebreton et al. [130] was unique in that the serum contained two deleted γ chains of different subclasses (IgG1 and IgG2). One of these subclasses persisted without change during the disease, and the level of the other subclass diminished and had disappeared at the time of death.
The reason for the relative predilection of γ-HCD to coexist with other, unrelated paraproteins is unclear. Three possible mechanisms have been delineated with bone marrow immunofluorescence studies (1) double, heavy-chain-specific precursor cells develop into two separate cell populations, each secreting different immunoglobulins of the same idiotype; (2) both paraproteins are synthesized simultaneously by cells of a single clone; and (3) rarely, two independent neoplastic cell lines coexist [131].
γ-Heavy-Chain Deposition Disease
Monoclonal immunoglobulin deposition disease is a well-recognized, pathologically defined entity. Continuous linear deposits of κ or, rarely, λ immunoglobulin light chains are the immunohistologic hallmark of light-chain deposition disease. Some cases of monoclonal immunoglobulin deposition disease have deposits of both light and heavy chains [132]. In 1992, Tubbs et al. [133] described two patients for whom they proposed the term pseudo γ-heavy-chain deposition disease. Both patients presented with acute renal failure. Renal biopsy showed a nodular intercapillary glomerulopathy and continuous, electron-dense granular deposits associated with a linear pattern of IgG4 heavy-chain deposition in vascular, tubular, and glomerular basement membranes. Light-chain deposits were absent in one patient and were faint and limited to the glomerular basement membrane in the other. The investigators believed that these immunohistologic findings were best explained by a change in the three-dimensional conformational structure of the protein after entrapment and binding to the basement membrane, rendering the light-chain antigenic sites inaccessible to antibody reagent and, thereby, undetectable. This hypothesis was the basis for their proposed designation of pseudo γ-heavy-chain deposition disease.
In view of the subsequent reports describing kidney deposits that contain short heavy chains but no detectable light chains [134, 135], the cases reported by Tubbs et al. [133] are now considered examples of γ-heavy-chain deposition disease (γ-HCDD). Since their report, not more than about two dozen documented cases of γ-HCDD have been reported in the medical literature. These cases have included the full subgroup spectrum of γ1 (seven patients) [136–140], γ2 (one patient) [141], γ3 (six patients) [41, 42, 140, 142–145], and γ4 (six patients) [133–135, 140, 146]. In several other patients with γ-HCDD, the subtype was not determined [140, 147–153].
In all cases of γ-HCDD in which the constant domains have been studied, the common feature is CH1 deletion [136]. In addition to this deletion, absence of the hinge region and CH2 domain has been reported in another patient [136]. The γ3 heavy chain from a patient with articular γ-HCDD was documented to start at the normal γ3 hinge region [154], similar to the findings for HCD protein γ3 OMM.
The most common clinical findings in γ-HCDD are nephrotic syndrome, hypertension, microhematuria, and, in some cases, hypocomplementemia [145]. Renal insufficiency is usually present at diagnosis. Beside renal involvement, there may be heavy-chain deposits in other organs, such as skin and skeletal muscle, as described in one case by Rott et al. [142], and in synovial tissue of a patient with seronegative rheumatoid arthritis, as described by Husby [42] and Husby et al. [41]. Cutis laxa associated with HCDD has been reported [146, 153].
The characteristic lesion on renal biopsy is nodular sclerosing glomerulopathy, sometimes with crescents. Immunofluorescence and electron microscopy show heavy-chain deposition in the mesangium and basement membranes of glomeruli, tubules, and blood vessels. Except for the composition of the deposits detected by immunofluorescence, the renal biopsy findings are indistinguishable from those of light-chain deposition disease and HCDD. In γ-HCDD, and in contrast to light-chain deposition disease, nodular glomerulosclerosis is a constant feature, hypertension and microhematuria are more frequent, and the hematologic disorder is generally mild. In most cases of γ-HCDD, a monoclonal protein can be documented in serum, urine, or bone marrow biopsy specimen, although its demonstration may require special studies. Often, the monoclonal proteins are detected in only minute quantities in serum or urine, probably in large part because of the avid tissue-binding properties of the heavy chains.
No consensus exists about the treatment of γ-HCDD, and in most cases the renal outcome is poor [144]. Moulin et al. [139] suggested that patients older than 60 years who have γ-HCDD in association with myeloma (which has been reported in four patients [134, 139, 144]) should be treated with conventional chemotherapy, but intensive therapy with peripheral blood stem-cell autografting should be considered for younger patients. Treatment of patients who have γ-HCDD without myeloma should depend on the clinical presentation. Five patients with γ-HCDD without associated multiple myeloma, reported by Lin et al. [140], received melphalan and prednisone (one patient), pulse dexamethasone (one patient), prednisone plus chlorambucil (one patient), or no treatment (two patients). Follow-up data showed that two patients had stable serum creatinine levels (over 5 months each) and three either had end-stage renal disease or needed immediate dialysis. One patient received a renal transplant from a living related donor and was doing well at 8 months posttransplant without recurrence of proteinuria. A patient reported by Soma et al. [145] was treated monthly with melphalan and prednisone, and ten courses of this therapy resulted in clinical remission [155]. The follow-up biopsy 2 years after onset showed remarkable diminution of both nodular glomerular lesions and IgG heavy-chain deposits in the mesangium, along the capillary walls, and on the tubular basement membrane [155]. In a patient reported by Herzenberg et al. [141], a renal transplant resulted in recurrent γ-HCDD in the transplanted organ after 2.5 years.
γ-Heavy-Chain-Associated Amyloidosis
Several heavy-chain-associated amyloid proteins have been described. Eulitz et al. [156] reported a patient (patient ART) with systemic heavy-chain amyloidosis in whom the amyloid component consisted of a short IgG heavy chain with a molecular weight of 22 kDa. The shortened heavy chain belonged to the γ1 subclass and contained a normal variable domain directly linked to the third constant domain. The amyloid protein (amyloid protein MAD) of another patient with heavy-chain-associated amyloidosis was characterized by the presence of a virtually intact VH region plus a D segment and lack of the JH segment and the entire CH region [157]. Table 36.2 summarizes the clinical data of seven patients with γ-heavy-chain-associated amyloidosis.
Table 36.2
Summary of clinical data for patients with γ-heavy-chain-associated amyloidosis
Age year/sex | Main clinical manifestation | Affected organ | Serum M protein | Plasma cell dyscrasia | Amyloid protein | VH subgroup | Reference No. |
|---|---|---|---|---|---|---|---|
65/F | Renal failure, hepatic failure | Kidney, liver, heart, spleen | IgGκ | + | γ Heavy chain (VH–CH3): 22 kDa | VH3 | [156] |
59/M | Nephrotic syndrome, renal failure | Kidney, spleen | IgGκ | + | γ Heavy chain (VH–D): 11 kDa | VH3 | [157] |
73/M | Proptosis | Orbit | Not detected | − | γ Heavy chain (CH3): 6 kDa | – | [158] |
53/F | Nephrotic syndrome | Kidney | IgGλ | + | γ Heavy chain | ND | [159] |
72/F | Renal failure | Kidney | IgGλ | + | γ Heavy chain (VH): 11 kDa | VH3 | [160] |
69/M | Pulmonary mass | Lung | Not detected | − (BLPD) | γ Heavy chain (VH): 12 and 18 kDa | ND | [161] |
61/F | Nephrotic syndrome | Kidney, gastrointestinal tract | IgGκ | − (BLPD) | γ Heavy chain (VH): 11 kDa | VH3 | [162] |
The molecular data are compatible with the hypothesis that constant-domain deletion in heavy chains may be responsible for free heavy-chain secretion, whereas variable-domain conformational singularities, rather than gross structural alterations, might promote either HCDD or heavy-chain-associated amyloidosis [136, 163, 164]. The discovery that certain forms of heavy chains, as well as light chains, can form amyloid provides further information on the chemical basis of amyloidogenicity and the diverse nature of this disease.
The diagnosis of heavy-chain-associated amyloidosis can be established with immunofluorescence staining of biopsy tissue with anti-heavy-chain antibodies. The clinical characteristics and course of heavy-chain-associated amyloidosis are similar to light-chain-associated amyloidosis. Katoh et al. [165] reported a patient with γ-heavy-chain-associated amyloidosis with an associated lymphoplasmacytic leukemia who was in a stable state with a nephrotic syndrome for 17 months since commencement of cyclic rituximab therapy.
α-Heavy-Chain Disease
In 1968, Seligmann et al. [166] described an Arab woman with severe malabsorption resulting from a lymphoplasmacytic infiltrate in the small bowel who had a monoclonal α-heavy chain in the serum. Since this first description, more than 400 cases [167] have been reported in the literature, and α-HCD is the most frequent of the HCDs. The disease is defined as a lymphoid proliferation involving the IgA secretory system and producing a homogeneous population of immunoglobulin molecules consisting of incomplete α chains devoid of light chains. The initial benign-appearing, antibiotic-responsive immunoproliferative lesions often evolve to fatal, highly malignant lymphoma. α-HCD might be considered a model showing the complex interactions of the environment with genetic factors and the complex infection–immunity–cancer interrelationships originating from the same proliferating clone.
Epidemiologic Factors
The syndrome of primary upper small intestinal lymphoma with malabsorption and clubbing of the fingers, which improved with small oral doses of tetracycline, was first described in Peru among poor mestizos [168, 169]. Most reports of α-HCD have been Arab or Jewish patients from the Mediterranean area or the Middle East, but numerous cases also have been described among inhabitants of Eastern Europe, the Indian subcontinent, the Far East, sub-Saharan Africa, and Central, North, and South America. Of interest, α-HCD in developed countries often occurs among immigrants from developing countries and in underprivileged native populations.
A common denominator for these patients is low socioeconomic status and substandard hygiene, resulting in recurrent infectious diarrhea and chronic parasitic infestation. Geophagia since early infancy was almost constant in patients at risk in Tunisia [170]. After studying the distribution of monoclonal gammopathies in Tunisia, the investigators published a report in 1990 showing that 17 % of 198 cases were attributed to α-HCD [171]. By comparison, a later study reported on 270 cases observed between 1992 and 2000 in the university hospital of Sfax, in which only 2.2 % were attributed to α-HCD [172], a finding that might be partially explained because of improved socioeconomic conditions.
Similarly, a persistent decrease in the incidence of immunoproliferative small intestinal disease (IPSID) since 1986 due to improvements in sanitation has been reported from Iran [173] and Greece [174]. Unlike multiple myeloma and the other HCDs, α-HCD has a predilection for young adults; most patients are in their twenties or thirties, although α-HCD has also been reported in children [175–180] and in persons in the seventh decade of life [181]. The prevalence of the disease is slightly higher in male persons than in female persons.
Pathogenesis
The cause of α-HCD is unknown. Current clinical, histologic, molecular, and immunologic data indicate that the cause of α-HCD is a complex, multistep process. The lymphoplasmacytic infiltration of the intestinal mucosa and regional mesenteric lymph nodes is likely a response of the alimentary tract’s immune system to protracted luminal antigenic stimulation. Bacterial lipopolysaccharides, dietary lectins [182–184], enterotoxins of Vibrio cholerae [185], oncogenic viruses [184, 186], and asbestosis [187] have been suspected of providing antigenic stimulation that triggers the histoimmunopathologic changes. The Epstein–Barr virus, which has been associated with B-cell lymphoproliferative disorders, was documented to have no role in the B-cell proliferation in IPSID of eight patients [188], whereas ultrastructural studies of lymph nodes of a patient with α-HCD, described by Arista-Nasr et al. [186], showed viruses that resembled the Epstein–Barr virus. A causal relationship between infection and pathogenesis is supported by the observation that α-HCD can respond to broad-spectrum antibiotics.
Using molecular strategies, Lecuit et al. [189] were able to detect Campylobacter jejuni in five of seven patients with α-HCD. However, no specific microorganism has been found in other clinical studies. The putative agent may be present only at the onset of the disease and absent at diagnosis.
The influence of environmental factors is supported by a report of spontaneous remission of α-HCD after the person with the disease left an endemic area [190] and a decline in the incidence rate of primary small intestinal lymphoma among Jewish persons born in Israel compared with Jewish immigrants with a relatively low socioeconomic standard from North Africa and Asia [191].
The postulated environmental antigenic stimulation might be associated with an underlying immunodeficiency, which could be due to malnutrition, especially in early infancy, or to genetic factors. An increased incidence of immunoglobulin abnormalities has been found in first-degree relatives of patients with α-HCD: 23 of 129 apparently healthy family members of eight patients with α-HCD had abnormal immunoglobulin patterns [192]. In the same families, the patients with α-HCD and the healthy first-degree relatives had an increase of circulating B lymphocytes and a decrease of T lymphocytes [193, 194]. They also had decreased cellular immunity, as shown by sensitization to dinitrochlorobenzene. Tuberculin skin test reactions were negative also.
A genetic element is suggested by the finding that patients with α-HCD have a greater association of HLA–AWI9 and HLA–BI2 antigens than do healthy blood donors or patients with malabsorption [195]. However, familial α-HCD has not been recognized.
Clinical Features
Most commonly, α-HCD presents as the digestive form. Its onset may be gradual or, more often, abrupt. During the early stage, diarrhea may be intermittent; progression of the disease is manifested by sustained chronic diarrhea, with malabsorption, steatorrhea, weight loss, abdominal pain, and vomiting [196]. Patients may present with abdominal surgical emergencies or chronic small-bowel obstruction [197]. “Tumoral” signs are more often observed in the late stages of the disease [198]. Ascites, tetany, or edema may be present. Amenorrhea, alopecia, and growth retardation in children and adolescents correlate with the duration and severity of the malabsorptive process [199]. Clubbing of the fingers appears to be more frequent than in any other intestinal disease [199]. Fever is uncommon. Hepatosplenomegaly and peripheral lymphadenopathy are infrequent findings also [200].
α-HCD may be confined to the respiratory tract, but this respiratory form is rare. Two of the described patients were children—an 8-year-old girl who presented with pulmonary infiltrates, hilar adenopathy, skull lesions, and a pharyngeal tumor [176] and a 3-year-old boy who had recurrent respiratory infections, hypogammaglobulinemia, and an α-HCD fragment [177]. Another patient presented with dyspnea and had diffuse interstitial pulmonary fibrosis, pleural effusion, and mediastinal nodes [201].
A lymphoma form of α-HCD has been described in three cases from Japan [202–205]. A striking clinical feature in two of these cases was long-standing and recurring skin eruptions, which developed before systemic lymphadenopathy [202, 203]. In the third patient, who had a history of rheumatoid arthritis, marked cervical and inguinal lymphadenopathy developed, and α-HCD protein was identified in serum and urine [204]. Infiltrating malignant cells in the lymph nodes were found to be the site of α-HCD protein synthesis. Neither the gastrointestinal nor the respiratory tract was involved in these patients.
Lymphomatous infiltration of the duodenum, jejunum, nasopharynx, and bone marrow was described in a Mauritanian man with α-HCD [206]. Also, α-HCD has been reported in a patient with a goiter from a plasmacytoma of the thyroid [207], in a patient with amyloidosis [208], and in a patient with polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin lesions [209].
Laboratory Findings
Hematologic and Metabolic Abnormalities
Mild to moderate anemia is often found in patients with α-HCD. Because the disease usually does not involve bone marrow, anemia occurs from malabsorption of folate, vitamin B12, and iron; dietary deficiency; or bleeding. Hypokalemia, hypocalcemia, and hypomagnesemia are common. The serum albumin level is nearly always low. The frequently increased serum alkaline phosphatase value is usually due to an increase in the intestinal isoenzyme fraction [210]. Serum lipid levels are low even when steatorrhea is mild. Results of the Schilling test with intrinsic factor are decreased in two-thirds of patients, and results of the d-xylose absorption test are almost always abnormal. The 24-h fecal fat excretion ranged between 6 and 15 g in 43 % and exceeded 15 g in 52 % of patients studied [196]. Heavy parasitic infestation of the intestine was a common occurrence; however, this occurrence did not appear to be substantially different from the general population living in the same area.
Immunologic Findings
Defects in humoral and delayed immunity have been shown in patients with α-HCD, even in the tumor-free stage [211]. An increase in circulating B lymphocytes and a decrease in T lymphocytes have been reported. Decreased cellular immunity has been shown with negative tuberculin skin tests and failure to be sensitized to dinitrochlorobenzene [192–194].
Radiographic Findings
Radiographic manifestations of α-HCD in the small intestine include hypertrophic and pseudopolypoid mucosal folds in the duodenum and jejunum that sometimes are associated with strictures or filling defects. These characteristics suggest extrinsic compression by hypertrophic peripancreatic or mesenteric lymph nodes [196]. Double contrast media studies of the small intestine are helpful in detecting precise mucosal changes [212].
Endoscopic Findings
Because α-HCD intestinal lesions nearly always affect the duodenum and jejunum, fiberoptic endoscopy with biopsies is a useful tool in the work-up of patients with suspected α-HCD. Five primary endoscopic patterns occurring either alone or in various combinations have been defined. The infiltrated pattern is the most specific finding, followed by the nodular pattern. Other primary lesions (ulcerations, mosaic pattern, and mucosal fold thickening alone) are nonspecific [213].
Protein Findings
Unlike in other monoclonal gammopathies, the characteristic sharp spike of a monoclonal protein is not seen on serum protein electrophoresis in α-HCD [214]. For about half of cases, an abnormal broad band is visible in the α2- or β-globulin region. This broad band is due to the propensity of α-heavy chains to form polymers. In the other patients, serum protein electrophoresis shows no evidence of an abnormal protein.
For most patients, the α-HCD protein can be found in the serum, but its level is often low [184]. Bence Jones proteinuria has never been documented. The level of α-HCD protein in the urine is low. In most cases studied, the α-HCD protein was also found in jejunal secretions when its presence was documented in the serum [170, 215]. Interestingly, however, α-HCD protein was found in the intestinal or gastric fluid in two cases in which it was undetectable in serum and urine [216, 217].
It is unclear whether monomeric or polymeric α-HCD protein is present in external secretions and whether α-HCD protein in jejunal fluid is linked to the secretory component. In several patients with α-HCD, no association was found between the α-HCD protein and the secretory component in the jejunal fluid or in the epithelial cells of the jejunum [178, 210, 218]. However, these data conflict with another report showing that jejunal fluid from two patients with α-HCD contained secretory component-associated α-HCD protein [219] and with immunofluorescence observations of rectal epithelial cells in an α-HCD case involving the large bowel [175, 220].
The linkage of polymeric α-HCD protein to the secretory component likely depends on balanced synthesis of α chains and J chains in the proliferating B cells, giving rise to polymers with the binding site for the secretory component expressed as an epithelial receptor. Insufficient receptor-mediated transport capacity results in passive external transfer of polymers without bound secretory component, along with leakage of serum-derived or locally produced monomeric α-HCD protein. The latter presumably originates from immunocytes with little or no J-chain synthesis [206].
In all cases studied so far, the α-HCD protein belonged to the α1 subclass. However, 10 % of normal serum immunoglobulin is of the α2 subclass.
Cytogenetic Abnormalities
Cytogenetic abnormalities have been found in the lymphoid cells of patients with α-HCD. The clonal proliferation in this disease appears to be associated with frequent alterations of chromosome 14 at band q32, resulting from translocations that differ from those observed in most other non-Hodgkin lymphomas [221]. Berger et al. [221] reported abnormal karyotypes in three of four patients, two of whom had not reached the stage of malignant lymphoma. In two instances, a rearrangement of 14q32 resulting from a t(9;14)(p11;q32) and a t(2;14)(p12;q32) translocation was observed. Cloning and sequencing of the der [14] break point of a chromosome translocation involving the 14q32 immunoglobulin locus suggested that the translocation originated from a local pairing of the chromosomes 9 and 14 [222, 223]. One case showed complex rearrangements, including t(5;9). No abnormalities were found in the intestinal tumor of the fourth patient with immunoblastic lymphoma. An abnormal chromosome marker (14q+) has been reported in the marrow of a patient with α-HCD [224].
Structural Protein Abnormalities
Most α-HCD proteins consist of multiple polymers. The molecular weight of the monomeric unit ranges from 29,000 to 34,000, and its length varies from one-half to three-fourths that of a normal α chain. The shortening results from an internal deletion involving most of VH and the first constant domain. Sequence data are available for several α-HCD proteins (Fig. 36.4). Common features of the defective α chain include deleted V regions, missing CH1 domains, and absence of light chains. Most of the proteins have short, nonimmunoglobulin-related sequences of unknown origin at the amino terminus. In all instances, the normal sequence of the α1 chain constant region resumes at the hinge. The complete sequences of the genes encoding three α-HCD proteins are shown in Fig. 36.5. These three genes show striking similarity in their position and extent of the two main deletions, which encompass sequences in the V/J and the switch/CH1 region.
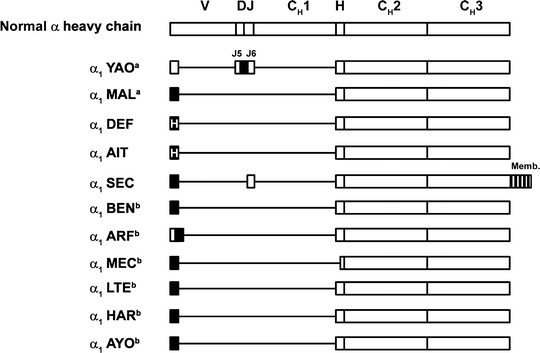
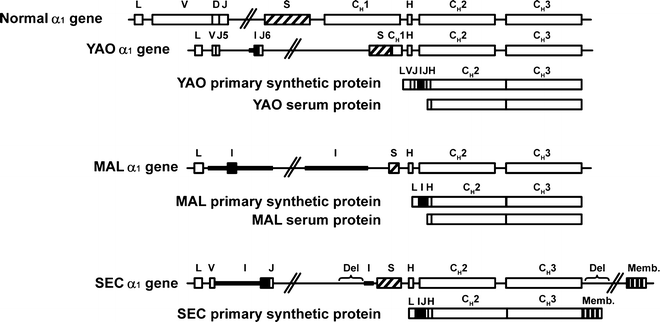
Fig. 36.4
Structure of various α-heavy-chain disease proteins compared with normal chain. indicates unusual amino acid sequences;
indicates unusual amino acid sequences;  , unusual and heterogeneous amino acid sequences; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; H hinge region; J joining region; lines, deletions; Memb membrane exon; V variable region. aStructures shown are primary synthetic products synthesized by the heavy-chain disease cells. Serum proteins were modified after synthesis and contained no amino acids before the hinge. bStructures shown are deduced amino acid sequences determined by cDNA sequencing. Citations for proteins are YAO [225], MAL [226], DEF [227], AIT [228], SEC [229], BEN [230], ARF [230], MEC [230], LTE [230], HAR [230], and AYO [230] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw Hill Companies.)
, unusual and heterogeneous amino acid sequences; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; H hinge region; J joining region; lines, deletions; Memb membrane exon; V variable region. aStructures shown are primary synthetic products synthesized by the heavy-chain disease cells. Serum proteins were modified after synthesis and contained no amino acids before the hinge. bStructures shown are deduced amino acid sequences determined by cDNA sequencing. Citations for proteins are YAO [225], MAL [226], DEF [227], AIT [228], SEC [229], BEN [230], ARF [230], MEC [230], LTE [230], HAR [230], and AYO [230] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw Hill Companies.)
indicates unusual amino acid sequences; , unusual and heterogeneous amino acid sequences; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; H hinge region; J joining region; lines, deletions; Memb membrane exon; V variable region. aStructures shown are primary synthetic products synthesized by the heavy-chain disease cells. Serum proteins were modified after synthesis and contained no amino acids before the hinge. bStructures shown are deduced amino acid sequences determined by cDNA sequencing. Citations for proteins are YAO [225], MAL [226], DEF [227], AIT [228], SEC [229], BEN [230], ARF [230], MEC [230], LTE [230], HAR [230], and AYO [230] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw Hill Companies.)Fig. 36.5
Structure of three genes coding for different α1-heavy-chain disease proteins compared with normal α1 gene.  indicates switch region;
indicates switch region; , inserted coding sequence;
, inserted coding sequence; , inserted noncoding sequence; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; Del deleted sequence; H hinge region; I inserted sequence; J joining region; L leader region; lines, intervening (noncoding) sequences; Memb membrane exon; S switch region; V variable region. Citations for proteins are YAO [225], MAL [226], and SEC [229] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)
, inserted noncoding sequence; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; Del deleted sequence; H hinge region; I inserted sequence; J joining region; L leader region; lines, intervening (noncoding) sequences; Memb membrane exon; S switch region; V variable region. Citations for proteins are YAO [225], MAL [226], and SEC [229] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)
indicates switch region;, inserted coding sequence;, inserted noncoding sequence; boxes, coding regions; CH1, CH2, and CH3, constant regions of heavy chains; D diversity segment; Del deleted sequence; H hinge region; I inserted sequence; J joining region; L leader region; lines, intervening (noncoding) sequences; Memb membrane exon; S switch region; V variable region. Citations for proteins are YAO [225], MAL [226], and SEC [229] (Adapted from Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal JT. Williams hematology. 7th edition. New York [NY]: McGraw-Hill; ©2006. Used with permission of The McGraw-Hill Companies.)Currently available molecular biologic studies indicate that genomic abnormalities, such as multiple deletion–insertion processes, mutations, or duplications that are focused in the VH–JH and CH1 regions, are at least partly responsible for the production of α-HCD proteins [231, 232]. These proteins are monoclonal even in the early stage of the disease [233, 234]. Although nonsecretory α-HCD has been described [92, 233], the molecular basis for nonsecretion is incompletely understood. In a case of nonsecretory α-HCD (α1-SEC), the productive α gene was noted to bear several noncontiguous deletions [229]. Two deletions were accompanied by peculiar insertions containing duplications. One of the deletions located at 3′ to CH3 eliminated the polyadenylation site of the secreted form of α-mRNA. As a result, only the membrane form of α-mRNA was present in the tumoral plasma cells, thus explaining the nonsecretory phenotype of the disease.
As a means to improved understanding about the molecular mechanism leading to the loss of light-chain production, a murine cell line of α-HCD was studied [235]. In this model, the failure of light-chain synthesis was shown to result from a disruption in the normal splicing pattern caused by the insertion of a 358-nucleotide nonimmunoglobulin sequence into the intron separating the leader exon from Vκ, leading to two mRNAs, neither of which encodes a functional light chain [235].
Pathologic Features
Enteric disease is the predominant manifestation of α-HCD. In this digestive form of α-HCD, the proliferation involves the whole length or at least the proximal half of the small intestine and the mesenteric lymph nodes. In a few cases, intestinal lesions spared the duodenum and jejunum or were limited to a segment of the latter [215]. Gastric and colorectal mucosae that belong to the IgA secretory system may be involved [217, 236]. α-HCD confined to the stomach [170, 237, 238] or presenting as a colonic mass [239] has been reported.
The disease progresses in three histopathologic stages, according to Galian et al. [240]. In stage A, a mature plasmacytic or lymphoplasmacytic infiltration of the mucosal lamina propria is noted. Villous atrophy is variable and inconstant. Stage B is characterized by the presence of atypical plasmacytic or lymphoplasmacytic cells and more or less atypical immunoblast-like cells extending to at least the submucosa. Subtotal or total villous atrophy is present. Stage C corresponds to an immunoblastic lymphoma, either forming discrete ulcerated tumors or extensively infiltrating long segments and invading the whole depth of the intestinal wall [240, 241]. Equivalent to the changes described in the small intestine, three histologic stages (A, B, and C) corresponding to the cellular type of infiltrate and the degree of the nodal architecture in the mesenteric lymph nodes have been described. Involvement of liver, spleen, and peripheral lymph nodes is uncommon.
The histologic lesions may progress at any given site from stage A to stage B or from stage B to stage C. However, different stages can be found at the same time in different organs or even at different sites of the same organ. This asynchronism is important for staging. Salem and Estephan [242] published a staging system based on the anatomical spread of α-HCD, which they suggested to be complementary to the Galian staging system; however, most use the Galian staging system for determining prognosis and therapeutic strategies.
The major lymphoma cell type in patients with α-HCD is immunoblastic lymphoma, with various degrees of plasmacytoid differentiation [170]. Although α-HCD associated with multiple polypoid lymphocytic lymphoma of the small intestine is rare, a patient with α-HCD associated with multiple polypoid lymphocytic lymphoma and leukemic manifestation without evidence of bone marrow involvement has been described [243]. The findings suggest that the circulating plasmacytoid lymphocytes originated from the tumor in the small intestine. Cytogenetic analysis showed the same abnormal karyotypes of neoplastic clones in the intestinal tumor cells as in the circulating leukemic cells.
Isaacson et al. [244] and Isaacson and Spencer [245, 246] suggested that the histopathologic findings in α-HCD fall into the group of lymphomas arising from mucosa-associated lymphoid tissue. Histologically, the diagnosis of mucosa-associated lymphoid tissue lymphoma is based on the existence of four elements: centrocyte-like cells, lymphoepithelial lesions, plasma cells, and reactive or residual follicles [247]. Spencer and Isaacson [246] hypothesized that in α-HCD, all large cells sometimes clustering in nodules at stage B are neoplastic follicular center cells, although they are often cytologically bizarre. Similarly, the invasion, disruption, and partial destruction of intestinal crypts, sometimes found even at stage A, are part of the lymphoepithelial lesions due to centrocyte-like cells of the same clonal origin as plasma cells and are pathognomonic of all gut-associated lymphoid tissue lymphomas [246]. Stage C tumors contain a mixture of the cytologic components [170, 215].
Lavergne et al. [248] described a case of α-HCD mimicking T-cell lymphoma. A marked predominance of small T cells in the mucosal infiltrate initially masked the underlying plasma cell proliferation, and small-T-cell lymphoma was diagnosed before immunoglobulin studies on the plasma cells were performed. The T cells were polyclonal, partially repressed during the follow-up period, and thought to be characteristic centrocyte-like cells.
In a few patients with the typical clinical and pathologic features of α-HCD, another monoclonal immunoglobulin (γ-HCD protein) [26, 27] a complete monoclonal IgA [249–252], or a polyclonal expression of IgA [253, 254] was found.
There has been some confusion about the terminology of so-called Mediterranean lymphoma. In 1976, the World Health Organization suggested the term IPSID [255]. This term should be restricted to small intestinal lesions whose pathologic factors are identical to those of α-HCD at any of its histologic stages irrespective of the type of immunoglobulin synthesized by the proliferating cells [256–258]. Because previously used methods to detect the protein were not very sensitive, data regarding the presence of the abnormal protein differ [259]. Rambaud et al. [170] reported that, among 19 consecutive patients with the epidemiologic, clinical, and pathologic features of IPSID, 16 had α-HCD protein in their serum and one had it in the jejunal secretion only. In one case, immunofluorescence study of the small bowel mucosa showed that most of the infiltrating cells were positive for α chains and negative for other heavy or light chains (nonsecretory) [217]. Patient 19 showed a massive infiltration of the small intestine by polyclonal plasma cells [253].
The pathologic changes in the few cases with the respiratory form of α-HCD are poorly documented. It can be postulated that additional cases of the respiratory form will be described in the future. In a case of lymph node form or lymphoma form, lymph node biopsy showed diffuse plasmacytic lymphoma [202].
Diagnosis
Because α-HCD in its intestinal form nearly always affects the duodenum and jejunum, endoscopy has been advocated as the first diagnostic procedure in the clinical investigation of patients in whom α-HCD is suspected. Enteric presentation of γ-HCD [26], monoclonal IgA secretion with a complete molecule [251], variable immunodeficiency [260], and acquired immunodeficiency syndrome [261] with clinicopathologic features simulating IPSID must be excluded.
The diagnosis of α-HCD depends on the identification of a free monoclonal α heavy chain. Several methods may be used to document α-HCD protein in biologic fluids [262]. A modified immunoselective technique described by Sun et al. [105] appears to be simple, convenient, and specific. The pathologic protein may easily escape detection by immunoelectrophoresis when its serum level is low. In most patients, the α-HCD protein can be found in the serum; in a few cases when it was undetectable in the serum, it could be documented in gastric fluid and jejunal secretions [217].
Synthesis of the α-HCD protein by the proliferating cells has been demonstrated through immunohistochemical or immunocytochemical methods and with biosynthesis studies in vitro. Use of these techniques is not necessary when the α-HCD protein is found in the serum or intestinal fluid, but they are helpful in the recognition of nonsecreting forms of α-HCD [217, 219, 229, 233, 263–266].
The quantity of abnormal α chains in the serum seems to be related to the nature (plasma-cell or immunoblastic type) of cells predominantly present in the intestinal mucosa or the mesenteric lymph nodes. During the disease course, the progressive diminution of mature plasma cells and their replacement by immature immunoblasts are followed by a progressive decrease in the serum concentration of α-HCD protein [267]. Hyposecretion of α-HCD protein may be found early and during the terminal evolutionary stage of the disease [268].
Treatment
In general, treatment depends on the extent and histopathologic stage of the disease. Because of the frequency of asynchronous histopathologic lesions, staging laparotomy should be performed [170, 269]. Supportive therapy with intravenously administered fluids, electrolytes, calcium and magnesium replacement, albumin, and, in some cases, total parenteral nutrition may be necessary in preparing the patient for staging laparotomy and during the early treatment period. Whether such improved diagnostic tools as computed tomography or magnetic resonance imaging will reduce the need for staging laparotomy remains to be seen. Given the predilection of α-HCD to occur in developing countries, it is unlikely that these diagnostic techniques will be available to most α-HCD patients.
Present therapeutic guidelines state that patients with stage A lesions limited to the bowel and mesenteric lymph nodes should be treated initially with oral antibiotics. In the case of intestinal bacterial overgrowth, antibiotics selected in accordance with the sensitivity pattern should be given. In the absence of a documented parasite or intestinal bacterial overgrowth, tetracycline or metronidazole and ampicillin are reasonable therapeutic choices. Any documented parasite should be eradicated. The eradication of Helicobacter pylori led to complete remission in two patients with α-HCD [270, 271], one of whom was unresponsive to prior combination chemotherapy [271]. In another patient with IPSID, eradication of H. pylori led to dramatic improvement of clinical, radiologic, and histologic parameters [272]. A trial of tetracycline therapy (1–2 g per day) for at least 6 months is the prerequisite for establishing responsiveness of the lesion [259, 269], although in cases where complete (chemical, immunologic, and histopathologic) remission is obtained with antibiotics alone, clinical improvement occurs early. Maintenance antibiotic treatment is unnecessary.
Close surveillance for early detection of overt lymphomatous transformation is advisable [273]. The development of abdominal lymphadenopathy and thickening of the small intestinal wall can be monitored with sequential ultrasonographic examinations [274]. Of interest, a persistently abnormal α-chain mRNA was found in one patient, despite an apparently complete clinical, pathologic, and immunopathologic remission after tetracycline therapy [233]. Subsequently, rapid recurrence of α-HCD developed, with transformation to immunoblastic lymphoma.
In patients with stage B or C disease, antiparasitic and antibiotic treatments are also useful for improving the malabsorption syndrome. Patients with stage B or C lesions or stage A lesions without marked improvement after 6 months of antibiotic treatment should be given chemotherapy. In a prospective randomized study, a doxorubicin-based regimen (Cytoxan [cyclophosphamide], Adriamycin [doxorubicin], Oncovin [vincristine], and prednisone) provided a higher response rate than either a protocol without doxorubicin (cyclophosphamide, vincristine, procarbazine, and prednisone) or total abdominal irradiation [275]. Similar results were noted in a retrospective study by Salimi and Spinelli [276]. Encouraging results were obtained in a treatment trial of cyclophosphamide, doxorubicin, teniposide, and prednisone with or without alternation with bleomycin, vinblastine, and doxorubicin [277]. Chemotherapy with cyclophosphamide, epidoxorubicin, vincristine, prednisolone, ifosfamide, methotrexate, VP-16, and dexamethasone resulted in complete remission in a patient with HCD associated with a high-grade, malignant non-Hodgkin lymphoma [278].
Surgical resection should be considered for focal or bulky transmural lymphomatous tumors. Because most patients with α-HCD are young, those with disseminated stage C disease showing good response to conventional or salvage chemotherapy could be candidates for autologous stem-cell transplant [279]. Previous trials have not incorporated immunotherapy with rituximab, an anti-CD20 monoclonal antibody, in the management of IPSID. As expected, the centrocyte-like cells are CD20 positive but the plasma cells are not. It would be interesting, in light of the extreme plasma cell differentiation and the plasmacytic nature of large-cell IPSID lymphoma, to investigate a possible role for newer multiple myeloma therapies, including proteasome inhibitors and immunomodulators [280].
Clinical Course
The course of α-HCD is variable but generally progressive in the absence of therapy. Follow-up care should include a periodic search for α-HCD protein in serum and urine and, if results are negative for the protein, in intestinal secretions. Bowel radiography, ultrasonography, and esophagogastroduodenojejunal endoscopy should be performed. A second-look laparotomy may be necessary [170].
Relapses may occur after treatment at any stage of the disease. Because antibiotic therapy in the early stage of intestinal α-HCD can result in full clinical remission, awareness of α-HCD and increased efforts to detect the disease before the lymphomatous phase are important [273, 281–283]. The disease may well be eradicated without any medical intervention through improving the socioeconomic status of the underprivileged populations in underdeveloped countries [269]. The long-term prognosis of patients with α-HCD continues to be imprecise because of the lack of large series with prolonged follow-up.
Spontaneous clinical and immunologic remission in an Italian patient with the digestive form of α-HCD was reported after the patient’s departure from Libya [190]. The α-HCD protein disappeared after total thyroidectomy in another patient with α-HCD, who presented with a goiter from an extramedullary plasmacytoma [284].
In a prospective study [277], 20 of 21 Tunisian patients with α-HCD underwent laparotomy and their disease was staged according to the method of Galian et al. [240]. Of the 21 patients, six were classified as having stage A, two as stage B, and 13 as stage C. The patients with stage A were first treated with antibiotics alone. Two of them had complete responses that were persisting 42 and 55 months later, and the four patients in whom antibiotic therapy failed received chemotherapy, with subsequent therapeutic failures in all four and two deaths. For stages B and C, combination therapy that included doxorubicin led to nine complete remissions with one early relapse, and salvage chemotherapy led to one more complete remission. Survival of the total group was 90 % at 2 years and 67 % at 3 years. All patients alive beyond 3.5 years were disease free.
Akbulut et al. [285] reported 5-year treatment results of 23 Turkish patients with IPSID, including five with the secretory type. Seven patients had stage A disease and were treated with tetracycline for a median of 7 months; the other 16 patients (nine with stage B and seven with stage C) received combination chemotherapy (cyclophosphamide, vincristine, procarbazine, and prednisolone). The median follow-up period was 68 months. Among patients with stage A disease, tetracycline yielded a 71 % complete response and a 43 % disease-free survival rate. Of the 16 patients (69 %) with stage B or C disease who received the chemotherapy regimen, 11 achieved complete response and only two had a recurrence (disease-free survival rate, 56 %). The 5-year overall survival rate for the entire group was 70 %, and the 5-year disease-free survival rate for patients with a complete response was 75 %. However, the median overall survival rate for three patients with immunoblastic lymphoma was only 7 months.
Price [286] studied 13 patients who had IPSID associated with α-HCD. Six patients—two with high-grade lymphoma and four with low-grade disease—received chemotherapy or radiation therapy, or both. One of these patients died at 76 months, and five were alive (three were disease free) an average of 92 months after presentation. Five patients, all with low-grade disease, received conservative therapy (antibiotics and, in some cases, prednisone and total parenteral nutrition). All were alive an average of 40 months after presentation. Three of these five patients achieved histologic remission at 5, 27, and 6 months, respectively. The other two of the five patients had persistent disease at 25 and 20 months, despite good clinical response. Two patients were not treated and died of high-grade lymphoma.
Shih et al. [287] described six patients who had α-HCD with lymphoma, mainly localized in the jejunum and the mesenteric nodes. The histologic subtypes were diffuse large cell for two patients, immunoblastic for three, and diffuse mixed for one. All patients responded poorly to chemotherapy (median survival, 10.5 months).
Malik et al. [288] studied 12 patients with IPSID, six of whom presented with stage A disease. Four patients responded to antibiotics or corticosteroids. In two patients, stage A disease evolved into stage C. One patient was lost to follow-up evaluation, and one patient was alive with disease. Of three patients who presented with stage B disease, two had complete response to chemotherapy, but the other patient refused treatment and died after 16 months. Three patients with stage C disease at diagnosis received aggressive combination chemotherapy and continued to have complete remission (median follow-up period, 2.2 years).
Manousos et al. [289] reported a patient with α-HCD who, after treatment for stage A disease (antibiotics and cyclophosphamide), achieved a complete remission lasting 18 months. Despite continued serologic remission (cured case of α-HCD), the patient had recurrent lymphoplasmacytic tumors of the small intestine, which were eventually completely eradicated through long-standing treatment with cyclophosphamide, vincristine, and prednisolone. Thorough investigation showed no evidence of α-HCD or lymphoma during a follow-up period of 26 years.
Preliminary results suggest that flow cytometric analysis of the S-phase fraction [290] and syndecan1, bcl6, and p53 [291] may be useful as a prognostic indicator and in the clinical treatment of patients with IPSID. Evaluation of the optimal treatment based on the literature is difficult, partly because of the small number of cases in any one study but mainly because of the poor long-term follow-up evaluation in most series. Because of the rarity of the disease, precise therapeutic protocols performed as multicenter studies are needed.
α-Heavy-Chain Deposition Disease
Three patients with α-HCDD have been reported in the medical literature. Cheng et al. [292] described a patient who presented with many of the clinicopathologic features common to patients with γ-HCDD—namely, hypertension, progressive renal failure, and nephrotic syndrome with renal biopsy showing crescentic nodular glomerulosclerosis and refractile granular electron-dense deposits in the glomerular and tubular basement membranes. The immune deposits stained for α-immunoglobulin heavy chain only but not for γ- and μ-immunoglobulin heavy chains and light chains and not with anti-α1 and anti-α2 subclass-specific reagents. On the basis of these findings, the investigators hypothesized that abnormally short α-immunoglobulin heavy chain may arise from a genetic mutation that deletes the genomic sequences that encode the CH1 and CH2 domains, similar to the findings in patients with γ-HCDD. Lin et al. [140] described a patient with α-HCDD who met the criteria for multiple myeloma. The third patient, reported by Chauveau et al. [293], presented with renal and skin deposits of a CH1-deleted α1 heavy chain.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


