Hallmarks of Cancer: An Organizing Principle for Cancer Medicine
Douglas Hanahan
Robert A. Weinberg
INTRODUCTION
The hallmarks of cancer comprise eight biologic capabilities acquired by incipient cancer cells during the multistep development of human tumors. The hallmarks constitute an organizing principle for rationalizing the complexities of neoplastic disease. They include sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprogramming energy metabolism, and evading immune destruction. Facilitating the acquisition of these hallmark capabilities are genome instability, which enables mutational alteration of hallmark-enabling genes, and immune inflammation, which fosters the acquisition of multiple hallmark functions. In addition to cancer cells, tumors exhibit another dimension of complexity: They contain a repertoire of recruited, ostensibly normal cells that contribute to the acquisition of hallmark traits by creating the tumor microenvironment. Recognition of the widespread applicability of these concepts will increasingly influence the development of new means to treat human cancer.
At the beginning of the new millennium, we proposed that six hallmarks of cancer embody an organizing principle that provides a logical framework for understanding the remarkable diversity of neoplastic diseases.1 Implicit in our discussion was the notion that, as normal cells evolve progressively to a neoplastic state, they acquire a succession of these hallmark capabilities, and that the multistep process of human tumor pathogenesis can be rationalized by the need of incipient cancer cells to acquire the diverse traits that in aggregate enable them to become tumorigenic and, ultimately, malignant.
We noted as an ancillary proposition that tumors are more than insular masses of proliferating cancer cells. Instead, they are complex tissues composed of multiple distinct types of neoplastic and normal cells that participate in heterotypic interactions with one another. We depicted the recruited normal cells, which form tumor-associated stroma, as active participants in tumorigenesis rather than passive bystanders; as such, these stromal cells contribute to the development and expression of certain hallmark capabilities. This notion has been solidified and extended during the intervening period, and it is now clear that the biology of tumors can no longer be understood simply by enumerating the traits of the cancer cells, but instead must encompass the contributions of the tumor microenvironment to tumorigenesis. In 2011, we revisited the original hallmarks, adding two new ones to the roster, and expanded on the functional roles and contributions made by recruited stromal cells to tumor biology.2 Herein we reiterate and further refine the hallmarks-of-cancer perspectives we presented in 2000 and 2011, with the goal of informing students of cancer medicine about the concept and its potential utility for understanding the pathogenesis of human cancer, and the potential relevance of this concept to the development of more effective treatments for this disease.
HALLMARK CAPABILITIES, IN ESSENCE
The eight hallmarks of cancer—distinct and complementary capabilities that enable tumor growth and metastatic dissemination—continue to provide a solid foundation for understanding the biology of cancer (Fig. 2.1). The sections that follow summarize the essence of each hallmark, providing insights into their regulation and functional manifestations.
Sustaining Proliferative Signaling
Arguably, the most fundamental trait of cancer cells involves their ability to sustain chronic proliferation. Normal tissues carefully control the production and release of growth-promoting signals that instruct entry of cells into and progression through the growthand-division cycle, thereby ensuring proper control of cell number and thus maintenance of normal tissue architecture and function. Cancer cells, by deregulating these signals, become masters of their own destinies. The enabling signals are conveyed in large part by growth factors that bind cell-surface receptors, typically containing intracellular tyrosine kinase domains. The latter proceed to emit signals via branched intracellular signaling pathways that regulate progression through the cell cycle as well as cell growth (that is, increase in cell size); often, these signals influence yet other cell-biologic properties, such as cell survival and energy metabolism.
Remarkably, the precise identities and sources of the proliferative signals operating within normal tissues remain poorly understood. Moreover, we still know relatively little about the mechanisms controlling the release of these mitogenic signals. In part, the study of these mechanisms is complicated by the fact that the growth factor signals controlling cell number and position within normal tissues are thought to be transmitted in a temporally and spatially regulated fashion from one cell to its neighbors; such paracrine signaling is difficult to access experimentally. In addition, the bioavailability of growth factors is regulated by their sequestration in the pericellular space and associated extracellular matrix. Moreover, the actions of these extracellular mitogenic proteins is further controlled by a complex network of proteases, sulfatases, and possibly other enzymes that liberate and activate these factors, apparently in a highly specific and localized fashion.
The mitogenic signaling operating in cancer cells is, in contrast, far better understood.3,4,5,6 Cancer cells can acquire the capability to sustain proliferative signaling in a number of alternative ways: They may produce growth factor ligands themselves, to which they can then respond via the coexpression of cognate receptors, resulting in autocrine proliferative stimulation. Alternatively, cancer cells may send signals to stimulate normal cells within the supporting tumor-associated stroma; the stromal cells then reciprocate by supplying the cancer cells with various growth factors.7,8 Mitogenic signaling can also be deregulated by elevating the levels of receptor proteins displayed at the cancer cell
surface, rendering such cells hyperresponsive to otherwise limiting amounts of growth factor ligands; the same outcome can result from structural alterations in the receptor molecules that facilitate ligand-independent firing.
surface, rendering such cells hyperresponsive to otherwise limiting amounts of growth factor ligands; the same outcome can result from structural alterations in the receptor molecules that facilitate ligand-independent firing.
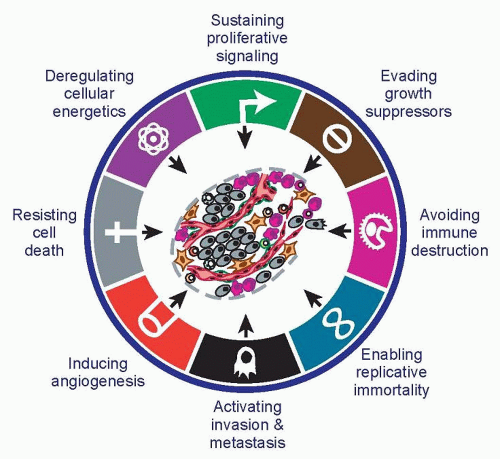 Figure 2.1 The hallmarks of cancer. Eight functional capabilities—the hallmarks of cancer—are thought to be acquired by developing cancers in the course of the multistep carcinogenesis that leads to most forms of human cancer. The order in which these hallmark capabilities are acquired and the relative balance and importance of their contributions to malignant disease appears to vary across the spectrum of human cancers. (Adapted from Hanahan D, Weinberg R. The hallmarks of cancer. Cell 2000;100:57-70; Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-674.) |
Independence from externally supplied growth factors may also derive from the constitutive activation of components of intracellular signaling cascades operating downstream of these receptors within cancer cells. These intracellular alterations obviate the need to stimulate cell proliferation pathways by ligand-mediated activation of cell-surface receptors. Of note, because a number of distinct downstream signaling pathways radiate from ligandstimulated receptors, the activation of one or another of these downstream branches (e.g., the pathway responding to the Ras signal transducer) may only provide a subset of the regulatory instructions transmitted by a ligand-activated receptor.
Somatic Mutations Activate Additional Downstream Pathways
DNA sequencing analyses of cancer cell genomes have revealed somatic mutations in certain human tumors that predict constitutive activation of the signaling circuits, cited previously, that are normally triggered by activated growth factor receptors. The past 3 decades have witnessed the identification in tens of thousands of human tumors of mutant, oncogenic alleles of the RAS protooncogenes, most of which have sustained point mutations in the 12th codon, which results in RAS proteins that are constitutively active in downstream signaling. Thus, more than 90% of pancreatic adenocarcinomas carry mutant K-RAS alleles. More recently, the repertoire of frequently mutated genes has been expanded to include those encoding the downstream effectors of the RAS proteins. For example, we now know that ˜40% of human melanomas contain activating mutations affecting the structure of the B-RAF protein, resulting in constitutive signaling through the RAF to the mitogen-activated protein (MAP)-kinase pathway.9 Similarly, mutations in the catalytic subunit of phosphoinositide 3-kinase (PI3K) isoforms are being detected in an array of tumor types; these mutations typically serve to hyperactivate the PI3K signaling pathway, causing in turn, excess signaling through the crucial Akt/PKB signal transducer.10,11 The advantages to tumor cells of activating upstream (receptor) versus downstream (transducer) signaling remain obscure, as does the functional impact of cross-talk between the multiple branched pathways radiating from individual growth factor receptors.
Disruptions of Negative-Feedback Mechanisms that Attenuate Proliferative Signaling
Recent observations have also highlighted the importance of negative-feedback loops that normally operate to dampen various types of signaling and thereby ensure homeostatic regulation of the flux of signals coursing through the intracellular circuitry.12,13,14,15 Defects in these negative-feedback mechanisms are capable of enhancing proliferative signaling. The prototype of this type of regulation involves the RAS oncoprotein. The oncogenic effects of mutant RAS proteins do not result from a hyperactivation of its downstream signaling powers; instead, the oncogenic mutations affecting RAS genes impair the intrinsic GTPase activity of RAS that normally serves to turn its activity off, ensuring that active signal transmission (e.g., from upstream growth factor receptors) is transient; as such, oncogenic RAS mutations disrupt an autoregulatory negative-feedback mechanism, without which RAS generates chronic proliferative signals.
Analogous negative-feedback mechanisms operate at multiple nodes within the proliferative signaling circuitry. A prominent example involves phosphatase and tensin homolog (PTEN), which counteracts PI3K by degrading its product, phosphatidylinositol 3,4,5-phosphate (PIP3). Loss-of-function mutations in PTEN amplify PI3K signaling and promote tumorigenesis in a variety of experimental models of cancer; in human tumors, PTEN expression is often lost by the methylation of DNA at specific sites associated with the promoter of the PTEN gene, resulting in the shutdown of its transcription.10,11
Yet another example involves the mammalian target of rapamycin (mTOR) kinase, a key coordinator of cell growth and metabolism that lies both upstream and downstream of the PI3K pathway. In the circuitry of some cancer cells, mTOR activation results, via negative feedback, in the inhibition of PI3K signaling. Accordingly, when mTOR is pharmacologically inhibited in such cancer cells (e.g., by the drug rapamycin), the associated loss of negative feedback results in increased activity of PI3K and its effector, the Akt/PKB kinase, thereby blunting the antiproliferative effects of mTOR inhibition.16,17 It is likely that compromised negative feedback loops in this and other signaling pathways will prove to be widespread among human cancer cells, serving as important means by which cancer cells acquire the capability of signaling chronically through these pathways. Moreover, disruption of such normally self-attenuating signaling can contribute to the development of adaptive resistance toward therapeutic drugs targeting mitogenic signaling.
Excessive Proliferative Signaling Can Trigger Cell Senescence
Early studies of oncogene action encouraged the notion that everincreasing expression of such genes and the signals released by their protein products would result in proportionately increased cancer cell proliferation and, thus, tumor growth. More recent research has undermined this notion, in that it is now apparent that excessively elevated signaling by oncoproteins, such as RAS, MYC, and RAF, can provoke counteracting (protective) responses from cells, such as induction of cell death; alternatively, cancer cells expressing high levels of these oncoproteins may be forced to enter into the nonproliferative but viable state called senescence. These responses contrast with those seen in cells expressing lower levels of these proteins, which permit cells to avoid senescence or cell death and, thus, proliferate.18,19,20,21
Cells with morphologic features of senescence, including enlarged cytoplasm, the absence of proliferation markers, and the expression of the senescence-induced β-galactosidase enzyme, are abundant in the tissues of mice whose genomes have been reengineered to cause overexpression of certain oncogenes19,20; such senescent cells are also prevalent in some cases of human melanoma.22
These ostensibly paradoxical responses seem to reflect intrinsic cellular defense mechanisms designed to eliminate cells experiencing excessive levels of certain types of mitogenic signaling. Accordingly, the intensity of oncogenic signaling observed in naturally arising cancer cells may represent compromises between maximal mitogenic stimulation and avoidance of these anti-proliferative defenses. Alternatively, some cancer cells may adapt to high levels of oncogenic signaling by disabling their senescence- or apoptosis-inducing circuitry.
Evading Growth Suppressors
In addition to the hallmark capability of inducing and sustaining positively acting growth-stimulatory signals, cancer cells must also circumvent powerful programs that negatively regulate cell proliferation; many of these programs depend on the actions of tumor suppressor genes. Dozens of tumor suppressors that operate in various ways to limit cell proliferation or survival have been discovered through their inactivation in one or another form of animal or human cancer; many of these genes have been validated as bona fide tumor suppressors through gain- or loss-of-function experiments in mice. The two prototypical tumor suppressor genes encode the retinoblastoma (RB)-associated and TP53 proteins; they operate as central control nodes within two key, complementary cellular regulatory circuits that govern the decisions of cells to proliferate, or alternatively, to activate growth arrest, senescence, or the cell-suicide program known as apoptosis.
The RB protein integrates signals from diverse extracellular and intracellular sources and, in response, decides whether or not a cell should proceed through its growth-and-division cycle.23,24,25 Cancer cells with defects in the RB pathway function are thus missing the services of a critical gatekeeper of cell-cycle progression whose absence permits persistent cell proliferation. Whereas RB transduces growth-inhibitory signals that largely originate outside of the cell, TP53 receives inputs from stress and abnormality sensors that function within the cell’s intracellular operating systems. For example, if the degree of damage to a cell’s genome is excessive, or if the levels of nucleotide pools, growth-promoting signals, glucose, or oxygenation are insufficient, TP53 can call a halt to further cell-cycle progression until these conditions have been normalized. Alternatively, in the face of alarm signals indicating overwhelming or irreparable damage to such cellular systems, TP53 can trigger apoptosis. Of note, the alternative effects of activated TP53 are complex and highly context dependent, varying by cell type as well as by the severity and persistence of conditions of cell-physiologic stress and genomic damage.
Although the two canonical suppressors of proliferation—TP53 and RB—have preeminent importance in regulating cell proliferation, various lines of evidence indicate that each operates as part of a larger network that is wired for functional redundancy. For example, chimeric mice populated throughout their bodies with individual cells lacking a functional Rb gene are surprisingly free of proliferative abnormalities, despite the expectation that a loss of RB function should result in unimpeded advance through the cell division cycle by these cells and their lineal descendants; some of the resulting clusters of Rb-null cells should, by all rights, progress to neoplasia. Instead, the Rb-null cells in such chimeric mice have been found to participate in relatively normal tissue morphogenesis throughout the body; the only neoplasia observed is of pituitary tumors developing late in life.26 Similarly, TP53-null mice develop normally, show largely normal cell and tissue homeostasis, and again develop abnormalities only later in life in the form of leukemias and sarcomas.27
Mechanisms of Contact Inhibition and Its Evasion
Four decades of research have demonstrated that the cell-to-cell contacts formed by dense populations of normal cells growing in 2-dimensional culture operate to suppress further cell proliferation, yielding confluent cell monolayers. Importantly, such contact inhibition is abolished in various types of cancer cells in culture, suggesting that contact inhibition is an in vitro surrogate of a mechanism that operates in vivo to ensure normal tissue homeostasis that is abrogated during the course of tumorigenesis. Until recently, the mechanistic basis for this mode of growth control remained obscure. Now, however, mechanisms of contact inhibition are beginning to emerge.28
One mechanism involves the product of the NF2 gene, long implicated as a tumor suppressor because its loss triggers a form of human neurofibromatosis. Merlin, the cytoplasmic NF2 gene product, orchestrates contact inhibition by coupling cell-surface adhesion molecules (e.g., E-cadherin) to transmembrane receptor tyrosine kinases (e.g., the EGF receptor). In so doing, Merlin strengthens the adhesiveness of cadherin-mediated cell-to-cell attachments. Additionally, by sequestering such growth factor receptors, Merlin limits their ability to efficiently emit mitogenic signals.28,29,30,31
Corruption of the TGF-β Pathway Promotes Malignancy
Transforming growth factor beta (TGF-β is best known for its antiproliferative effects on epithelial cells. The responses of carcinoma cells to TGF-β’s proliferation-suppressive effects is now appreciated to be far more elaborate than a simple shutdown of its signaling circuitry.32,33,34,35 In normal cells, exposure to TGF-β blocks their progression through the G1 phase of the cell cycle. In many latestage tumors, however, TGF-β signaling is redirected away from suppressing cell proliferation and is found instead to activate a cellular program, termed the epithelial-to-mesenchymal transition (EMT), which confers on cancer cells multiple traits associated with high-grade malignancy, as will be discussed in further detail.
Resisting Cell Death
The ability to activate the normally latent apoptotic cell-death program appears to be associated with most types of normal cells throughout the body. Its actions in many if not all multicellular organisms seems to reflect the need to eliminate aberrant cells whose continued presence would otherwise threaten organismic integrity. This rationale explains why cancer cells often, if not invariably, inactivate or attenuate this program during their development.21,36,37,38
Elucidation of the detailed design of the signaling circuitry governing the apoptotic program has revealed how apoptosis is triggered in response to various physiologic stresses that cancer cells experience either during the course of tumorigenesis or as a result of anticancer therapy. Notable among the apoptosis-inducing stresses are signaling imbalances resulting from elevated levels of oncogene signaling and from DNA damage. The regulators of the apoptotic response are divided into two major circuits, one receiving and processing extracellular death-inducing signals (the extrinsic apoptotic program, involving for example the Fas ligand/Fas receptor), and the other sensing and integrating a variety of signals of intracellular origin (the intrinsic program). Each of these circuits culminates in the activation of a normally latent protease (caspase 8 or 9, respectively), which proceeds to initiate a cascade of proteolysis involving effector caspases that are responsible for the execution phase of apoptosis. During this final phase, an apoptotic
cell is progressively disassembled and then consumed, both by its neighbors and by professional phagocytic cells. Currently, the intrinsic apoptotic program is more widely implicated as a barrier to cancer pathogenesis.
cell is progressively disassembled and then consumed, both by its neighbors and by professional phagocytic cells. Currently, the intrinsic apoptotic program is more widely implicated as a barrier to cancer pathogenesis.
The molecular machinery that conveys signals between the apoptotic regulators and effectors is controlled by counterbalancing pro- and antiapoptotic members of the Bcl-2 family of regulatory proteins.36,37 The archetype, Bcl-2, along with its closest relatives (Bcl-XL, Bcl-W, Mcl-1, A1) are inhibitors of apoptosis, acting in large part by binding to and thereby suppressing two proapoptotic triggering proteins (Bax and Bak); the latter are embedded in the mitochondrial outer membrane. When relieved of inhibition by their antiapoptotic relatives, Bax and Bax disrupt the integrity of the outer mitochondrial membrane, causing the release into the cytosol of proapoptotic signaling proteins, the most important of which is cytochrome C. When the normally sequestered cytochrome C is released, it activates a cascade of cytosolic caspase proteases that proceed to fragment multiple cellular structures, thereby executing the apoptotic death program.37,39
Several abnormality sensors have been identified that play key roles in triggering apoptosis.21,37 Most notable is a DNA damage sensor that acts through the TP53 tumor suppressor40; TP53 induces apoptosis by upregulating expression of the proapoptotic, Bcl-2-related Noxa and Puma proteins, doing so in response to substantial levels of DNA breaks and other chromosomal abnormalities. Alternatively, insufficient survival factor signaling (e.g., inadequate levels of interleukin (IL)-3 in lymphocytes or of insulinlike growth factors 1/2 [IGF1/2] in epithelial cells) can elicit apoptosis through another proapoptotic Bcl-2-related protein called Bim. Yet another condition triggering apoptosis involves hyperactive signaling by certain oncoproteins, such as Myc, which acts in part via Bim and other Bcl-2-related proteins.18,21,40
Tumor cells evolve a variety of strategies to limit or circumvent apoptosis. Most common is the loss of TP53 tumor suppressor function, which eliminates this critical damage sensor from the apoptosis-inducing circuitry. Alternatively, tumors may achieve similar ends by increasing the expression of antiapoptotic regulators (Bcl-2, Bcl-XL) or of survival signals (IGF1/2), by downregulating proapoptotic Bcl-2-related factors (Bax, Bim, Puma), or by short-circuiting the extrinsic ligand-induced death pathway. The multiplicity of apoptosis-avoiding mechanisms presumably reflects the diversity of apoptosis-inducing signals that cancer cell populations encounter during their evolution from the normal to the neoplastic state.
Autophagy Mediates Both Tumor Cell Survival and Death
Autophagy represents an important cell-physiologic response that, like apoptosis, normally operates at low, basal levels in cells but can be strongly induced in certain states of cellular stress, the most obvious of which is nutrient deficiency.41,42,43 The autophagic program enables cells to break down cellular organelles, such as ribosomes and mitochondria, allowing the resulting catabolites to be recycled and thus used for biosynthesis and energy metabolism. As part of this program, intracellular vesicles (termed autophagosomes) envelope the cellular organelles destined for degradation; the resulting vesicles then fuse with lysosomes in which degradation occurs. In this fashion, low-molecular-weight metabolites are generated that support survival in the stressed, nutrient-limited environments experienced by many cancer cells. When acting in this fashion, autophagy favors cancer cell survival.
However, the autophagy program intersects in more complex ways with the life and death of cancer cells. Like apoptosis, the autophagy machinery has both regulatory and effector components.41,42,43 Among the latter are proteins that mediate autophagosome formation and delivery to lysosomes. Of note, recent research has revealed intersections between the regulatory circuits governing autophagy, apoptosis, and cellular homeostasis. For example, the signaling pathway involving PI3K, AKT, and mTOR, which is stimulated by survival signals to block apoptosis, similarly inhibits autophagy; when survival signals are insufficient, the PI3K signaling pathway is downregulated, with the result that autophagy and/or apoptosis may be induced.41,42,44,45
Another interconnection between these two programs resides in the Beclin-1 protein, which has been shown by genetic studies to be necessary for the induction of autophagy.41,42,43,44 Beclin-1 is a member of the Bcl-2 family of apoptotic regulatory proteins, and its BH3 domain allows it to bind the Bcl-2/Bcl-XL proteins. Stress sensor-coupled BH3-containing proteins (e.g. Bim, Noxa) can displace Beclin-1 from its association with Bcl-2/Bcl-XL, enabling the liberated Beclin-1 to trigger autophagy, much as they can release proapoptotic Bax and Bak to trigger apoptosis. Hence, stresstransducing Bcl-2-related proteins can induce apoptosis and/or autophagy depending on the physiologic state of the cell.
Genetically altered mice bearing inactivated alleles of the Beclin-1 gene or of certain other components of the autophagy machinery exhibit increased susceptibility to cancer.42,46 These results suggest that the induction of autophagy can serve as a barrier to tumorigenesis that may operate independently of or in concert with apoptosis. For example, excessive activation of the autophagy program may cause cells to devour too many of their own critical organelles, such that cell growth and division are crippled. Accordingly, autophagy may represent yet another barrier that needs to be circumvented by incipient cancer cells during multistep tumor development.41,46
Perhaps paradoxically, nutrient starvation, radiotherapy, and certain cytotoxic drugs can induce elevated levels of autophagy that apparently protect cancer cells.45,46,47,48 Moreover, severely stressed cancer cells have been shown to shrink via autophagy to a state of reversible dormancy.46,49 This particular survival response may enable the persistence and eventual regrowth of some latestage tumors following treatment with potent anticancer agents. Together, observations like these indicate that autophagy can have dichotomous effects on tumor cells and, thus, tumor progression.46,47 An important agenda for future research will involve clarifying the genetic and cell-physiologic conditions that determine when and how autophagy enables cancer cells to survive or, alternatively, causes them to die.
Necrosis Has Proinflammatory and Tumor-Promoting Potential
In contrast to apoptosis, in which a dying cell contracts into an almost invisible corpse that is soon consumed by its neighbors, necrotic cells become bloated and explode, releasing their contents into the local tissue microenvironment. A body of evidence has shown that cell death by necrosis, like apoptosis, is an organized process under genetic control, rather than being a random and undirected process.50,51,52
Importantly, necrotic cell death releases proinflammatory signals into the surrounding tissue microenvironment, in contrast to apoptosis, which does not. As a consequence, necrotic cells can recruit inflammatory cells of the immune system,51,53,54 whose dedicated function is to survey the extent of tissue damage and remove associated necrotic debris. In the context of neoplasia, however, multiple lines of evidence indicate that immune inflammatory cells can be actively tumor-promoting by fostering angiogenesis, cancer cell proliferation, and invasiveness (discussed in subsequent sections). Additionally, necrotic cells can release bioactive regulatory factors, such as IL1α, which can directly stimulate neighboring viable cells to proliferate, with the potential, once again, to facilitate neoplastic progression.53 Consequently, necrotic cell death, while seemingly beneficial in counterbalancing cancer-associated hyperproliferation, may ultimately do more damage to the patient than good.
Enabling Replicative Immortality
Cancer cells require unlimited replicative potential in order to generate macroscopic tumors. This capability stands in marked contrast to the behavior of the cells in most normal cell lineages in the body, which are only able to pass through a limited number of successive cell growth-and-division cycles. This limitation has been associated with two distinct barriers to proliferation: replicative senescence, a typically irreversible entrance into a nonproliferative but viable state, and crisis, which involves cell death. Accordingly, when cells are propagated in culture, repeated cycles of cell division lead first to induction of replicative senescence and then, for those cells that succeed in circumventing this barrier, to the crisis phase, in which the great majority of cells in the population die. On rare occasion, cells emerge from a population in crisis and exhibit unlimited replicative potential. This transition has been termed immortalization, a trait that most established cell lines possess by virtue of their ability to proliferate in culture without evidence of either senescence or crisis.
Multiple lines of evidence indicate that telomeres protecting the ends of chromosomes are centrally involved in the capability for unlimited proliferation.55,56,57,58 The telomere-associated DNA, composed of multiple tandem hexanucleotide repeats, shortens progressively in the chromosomes of nonimmortalized cells propagated in culture, eventually losing the ability to protect the ends of chromosomal DNA from end-to-end fusions; such aberrant fusions generate unstable dicentric chromosomes, whose resolution during the anaphase of mitosis results in a scrambling of karyotype and entrance into crisis that threatens cell viability. Accordingly, the length of telomeric DNA in a cell dictates how many successive cell generations its progeny can pass through before telomeres are largely eroded and have consequently lost their protective functions.
Telomerase, the specialized DNA polymerase that adds telomere repeat segments to the ends of telomeric DNA, is almost absent in nonimmortalized cells but is expressed at functionally significant levels in the great majority (˜90%) of spontaneously immortalized cells, including human cancer cells. By extending telomeric DNA, telomerase is able to counter the progressive telomere erosion that would otherwise occur in its absence. The presence of telomerase activity, either in spontaneously immortalized cells or in the context of cells engineered to express the enzyme, is correlated with a resistance to induction of both senescence and crisis/apoptosis; conversely, the suppression of telomerase activity leads to telomere shortening and to activation of one or the other of these proliferative barriers.
The two barriers to proliferation—replicative senescence and crisis/apoptosis—have been rationalized as crucial anticancer defenses that are hardwired into our cells and are deployed to impede the outgrowth of clones of preneoplastic and, frankly, neoplastic cells. According to this thinking, most incipient neoplasias exhaust their endowment of replicative doublings and are stopped in their tracks by either of these barriers. The eventual immortalization of rare variant cells that proceed to form tumors has been attributed to their ability to maintain telomeric DNA at lengths sufficient to avoid triggering either senescence or apoptosis, which is achieved most commonly by upregulating the expression of telomerase or, less frequently, via an alternative recombination-based (ALT) telomere maintenance mechanism.59 Hence, telomere shortening has come to be viewed as a clocking device that determines the limited replicative potential of normal cells and, thus, one that must be overcome by cancer cells.
Reassessing Replicative Senescence
The senescent state induced by oncogenes, as described previously, is remarkably similar to that induced when cells are explanted from living tissue and introduced into culture, the latter being the replicative senescence just discussed. Importantly, the concept of replication-induced senescence as a general barrier requires refinement and reformulation. Recent experiments have revealed that the induction of senescence in certain cultured cells can be delayed and possibly eliminated by the use of improved cell culture conditions, suggesting that recently explanted primary cells may be intrinsically able to proliferate unimpeded in culture up the point of crisis and the associated induction of apoptosis triggered by critically shortened telomeres.60,61,62,63 This result indicates that telomere shortening does not necessarily induce senescence prior to crisis. Additional insight comes from experiments in mice engineered to lack telomerase; this work has revealed that shortening telomeres can shunt premalignant cells into a senescent state that contributes (along with apoptosis) to attenuated tumorigenesis in mice genetically destined to develop particular forms of cancer.58 Such telomerase-null mice with highly eroded telomeres exhibit multiorgan dysfunction and abnormalities that provide evidence of both senescence and apoptosis, perhaps similar to the senescence and apoptosis observed in cell culture.58,64 Thus, depending on the cellular context, the proliferative barrier of telomere shortening can be manifested by the induction of senescence and/or apoptosis.
Delayed Activation of Telomerase May Both Limit and Foster Neoplastic Progression
There is now evidence that clones of incipient cancer cells in spontaneously arising tumors experience telomere loss-induced crisis relatively early during the course of multistep tumor progression due to their inability to express significant levels of telomerase. Thus, extensively eroded telomeres have been documented in premalignant growths through the use of fluorescence in situ hybridization (FISH), which has also revealed the end-to-end chromosomal fusions that signal telomere failure and crisis.65,66 These results suggest that such incipient cancer cells have passed through a substantial number of successive telomere-shortening cell divisions during their evolution from fully normal cells of origin.
Accordingly, the development of some human neoplasias may be aborted by telomere-induced crisis long before they have progressed to become macroscopic, frankly neoplastic growths. A quite different situation is observed in cells that have lost the TP53-mediated surveillance of genomic integrity and, thereafter, experience critically eroded telomeres. The loss of the TP53 DNA damage sensor can enable such cells to avoid apoptosis that would otherwise be triggered by the DNA damage resulting from dysfunctional telomeres. Instead, such cells lacking TP53 continue to divide, suffering repeated cycles of interchromosomal fusion and subsequent breakage at mitosis. Such breakage-fusion-bridge (BFB) cycles result in deletions and amplifications of chromosomal segments, evidently serving to mutagenize the genome, thereby facilitating the generation and subsequent clonal selection of cancer cells that have acquired mutant oncogenes and tumor suppressor genes.58,67 One infers, however, that the clones of cancer cells that survive this telomere collapse must eventually acquire the ability to stabilize and thus protect their telomeres via the activation of telomerase or the ALT mechanism noted previously.
These considerations present an interesting dichotomy: Although dysfunctional telomeres are an evident barrier to chronic proliferation, they can also facilitate the genomic instability that generates hallmark-enabling mutations, as will be discussed further. Both mechanisms may be at play in certain forms of carcinogenesis in the form of transitory telomere deficiency prior to telomere stabilization. Circumstantial support for this concept of transient telomere deficiency in facilitating malignant progression has come from comparative analyses of premalignant and malignant lesions in the human breast.68,69 The premalignant lesions did not express significant levels of telomerase and were marked by telomere shortening and chromosomal aberrations. In contrast, overt carcinomas exhibited telomerase expression concordantly with the reconstruction of longer telomeres and the fixation of the
aberrant karyotypes that would seem to have been acquired after telomere failure but before the acquisition of telomerase activity. When portrayed in this way, the delayed acquisition of telomerase function serves to generate tumor-promoting mutations, whereas its subsequent expression stabilizes the mutant genome and confers the unlimited replicative capacity that cancer cells require in order to generate clinically apparent tumors.
aberrant karyotypes that would seem to have been acquired after telomere failure but before the acquisition of telomerase activity. When portrayed in this way, the delayed acquisition of telomerase function serves to generate tumor-promoting mutations, whereas its subsequent expression stabilizes the mutant genome and confers the unlimited replicative capacity that cancer cells require in order to generate clinically apparent tumors.
Inducing Angiogenesis
Like normal tissues, tumors require sustenance in the form of nutrients and oxygen as well as an ability to evacuate metabolic wastes and carbon dioxide. The tumor-associated neovasculature, generated by the process of angiogenesis, addresses these needs. During embryogenesis, the development of the vasculature involves the birth of new endothelial cells and their assembly into tubes (vasculogenesis) in addition to the sprouting (angiogenesis) of new vessels from existing ones. Following this morphogenesis, the normal vasculature becomes largely quiescent. In the adult, as part of physiologic processes such as wound healing and female reproductive cycling, angiogenesis is turned on, but only transiently. In contrast, during tumor progression, an angiogenic switch is almost always activated and remains on, causing normally quiescent vasculature to continually sprout new vessels that help sustain expanding neoplastic growths.70
A compelling body of evidence indicates that the angiogenic switch is governed by countervailing factors that either induce or oppose angiogenesis.71,72 Some of these angiogenic regulators are signaling proteins that bind to stimulatory or inhibitory cell-surface receptors displayed by vascular endothelial cells. The well-known prototypes of angiogenesis inducers and inhibitors are vascular endothelial growth factor-A (VEGF-A) and thrombospondin-1 (Tsp-1), respectively.
The VEGF-A gene encodes ligands that are involved in orchestrating new blood vessel growth during embryonic and postnatal development, in the survival of endothelial cells in already-formed vessels, and in certain physiologic and pathologic situations in the adult. VEGF signaling via three receptor tyrosine kinases (VEGFR1-3) is regulated at multiple levels, reflecting this complexity of purpose. VEGF gene expression can be upregulated both by hypoxia and by oncogene signaling.73,74,75 Additionally, VEGF ligands can be sequestered in the extracellular matrix in latent forms that are subject to release and activation by extracellular matrix-degrading proteases (e.g., matrix metallopeptidase 9 [MMP-9]).76 In addition, other proangiogenic proteins, such as members of the fibroblast growth factor (FGF) family, have been implicated in sustaining tumor angiogenesis.71 TSP-1, a key counterbalance in the angiogenic switch, also binds transmembrane receptors displayed by endothelial cells and thereby triggers suppressive signals that can counteract proangiogenic stimuli.77
The blood vessels produced within tumors by an unbalanced mix of proangiogenic signals are typically aberrant: Tumor neovasculature is marked by precocious capillary sprouting, convoluted and excessive vessel branching, distorted and enlarged vessels, erratic blood flow, microhemorrhaging, leaking of plasma into the tissue parenchyma, and abnormal levels of endothelial cell proliferation and apoptosis.78,79
Angiogenesis is induced surprisingly early during the multistage development of invasive cancers both in animal models and in humans. Histologic analyses of premalignant, noninvasive lesions, including dysplasias and in situ carcinomas arising in a variety of organs, have revealed the early tripping of the angiogenic switch.70,80 Historically, angiogenesis was envisioned to be important only when rapidly growing macroscopic tumors had formed, but more recent data indicate that angiogenesis also contributes to the microscopic premalignant phase of neoplastic progression, further cementing its status as an integral hallmark of cancer.
Gradations of the Angiogenic Switch
Once angiogenesis has been activated, tumors exhibit diverse patterns of neovascularization. Some tumors, including highly aggressive types such as pancreatic ductal adenocarcinomas, are hypovascularized and replete with stromal deserts that are largely avascular and indeed may even be actively antiangiogenic.81 In contrast, many other tumors, including human renal and pancreatic neuroendocrine carcinomas, are highly angiogenic and, consequently, densely vascularized.82,83
Collectively, such observations suggest an initial tripping of the angiogenic switch during tumor development, which is followed by a variable intensity of ongoing neovascularization, the latter being controlled by a complex biologic rheostat that involves both the cancer cells and the associated stromal microenvironment.71,72 Of note, the switching mechanisms can vary, even though the net result is a common inductive signal (e.g., VEGF). In some tumors, dominant oncogenes operating within tumor cells, such as Ras and Myc, can upregulate the expression of angiogenic factors, whereas in others, such inductive signals are produced indirectly by immune inflammatory cells, as will be discussed.
Endogenous Angiogenesis Inhibitors Present Natural Barriers to Tumor Angiogenesis
A variety of secreted proteins have been reported to have the capability to help shut off normally transitory angiogenesis, including thrombospondin-1 (TSP-1), fragments of plasmin (angiostatin) and type 18 collagen (endostatin), along with another dozen candidate antiangiogenic proteins.77,84,85,86,87,88 Most are proteins, and many are derived by proteolytic cleavage of structural proteins that are not themselves angiogenic regulators.
A number of these endogenous inhibitors of angiogenesis can be detected in the circulation of normal mice and humans. Genes that encode several endogenous angiogenesis inhibitors have been deleted from the mouse germ line without untoward developmental or physiologic effects; however, the growth of autochthonous and implanted tumors is enhanced as a consequence.84,85,88 By contrast, if the circulating levels of an endogenous inhibitor are genetically increased (e.g., via overexpression in transgenic mice or in xenotransplanted tumors), tumor growth is impaired.85,88 Interestingly, wound healing and fat deposition are impaired or accelerated by elevated or ablated expression of such genes.89,90 The data suggest that, under normal conditions, endogenous angiogenesis inhibitors serve as physiologic regulators modulating the transitory angiogenesis that occurs during tissue remodeling and wound healing; they may also act as intrinsic barriers to the induction and/or persistence of angiogenesis by incipient neoplasias.
Pericytes Are Important Components of the Tumor Neovasculature
Pericytes have long been known as supporting cells that are closely apposed to the outer surfaces of the endothelial tubes in normal tissue vasculature, where they provide important mechanical and physiologic support to the endothelial cells. Microscopic studies conducted in recent years have revealed that pericytes are associated, albeit loosely, with the neovasculature of most, if not all, tumors.91,92,93 More importantly, mechanistic studies (discussed subsequently) have revealed that pericyte coverage is important for the maintenance of a functional tumor neovasculature.
A Variety of Bone Marrow-Derived Cells Contribute to Tumor Angiogenesis
It is now clear that a repertoire of cell types originating in the bone marrow play crucial roles in pathologic angiogenesis.94,95,96,97 These include cells of the innate immune system—notably, macrophages, neutrophils, mast cells, and myeloid progenitors—that assemble
at the margins of such lesions or infiltrate deeply within them; the tumor-associated inflammatory cells can help to trip the angiogenic switch in quiescent tissue and sustain ongoing angiogenesis associated with tumor growth. In addition, they can help protect the vasculature from the effects of drugs targeting endothelial cell signaling.98 Moreover, several types of bone marrow-derived vascular progenitor cells have been observed to have migrated into neoplastic lesions and become intercalated into the existing neovasculature, where they assumed the roles of either pericytes or endothelial cells.92,99,100
at the margins of such lesions or infiltrate deeply within them; the tumor-associated inflammatory cells can help to trip the angiogenic switch in quiescent tissue and sustain ongoing angiogenesis associated with tumor growth. In addition, they can help protect the vasculature from the effects of drugs targeting endothelial cell signaling.98 Moreover, several types of bone marrow-derived vascular progenitor cells have been observed to have migrated into neoplastic lesions and become intercalated into the existing neovasculature, where they assumed the roles of either pericytes or endothelial cells.92,99,100
Activating Invasion and Metastasis
The multistep process of invasion and metastasis has been schematized as a sequence of discrete steps, often termed the invasion-metastasis cascade.101,102 This depiction portrays a succession of cell-biologic changes, beginning with local invasion, then intravasation by cancer cells into nearby blood and lymphatic vessels, transit of cancer cells through the lymphatic and hematogenous systems, followed by the escape of cancer cells from the lumina of such vessels into the parenchyma of distant tissues (extravasation), the formation of small nests of cancer cells (micrometastases), and finally, the growth of micrometastatic lesions into macroscopic tumors, this last step being termed colonization. These steps have largely been studied in the context of carcinoma pathogenesis. Indeed, when viewed through the prism of the invasion—metastasis cascade, the diverse tumors of this class appear to behave in similar ways.
During the malignant progression of carcinomas, the neoplastic cells typically develop alterations in their shape as well as their attachment to other cells and to the extracellular matrix (ECM). The best-characterized alteration involves the loss by carcinoma cells of E-cadherin, a key epithelial cell-to-cell adhesion molecule. By forming adherens junctions between adjacent epithelial cells, E-cadherin helps to assemble epithelial cell sheets and to maintain the quiescence of the cells within these sheets. Moreover, increased expression of E-cadherin has been well established as an antagonist of invasion and metastasis, whereas a reduction of its expression is known to potentiate these behaviors. The frequently observed downregulation and occasional mutational inactivation of the E-cadherin-encoding gene, CDH1, in human carcinomas provides strong support for its role as a key suppressor of the invasion-metastasis hallmark capability.103,104
Notably, the expression of genes encoding other cell-to-cell and cell-to-ECM adhesion molecules is also significantly altered in the cells of many highly aggressive carcinomas, with those favoring cytostasis typically being downregulated. Conversely, adhesion molecules normally associated with the cell migrations that occur during embryogenesis and inflammation are often upregulated. For example, N-cadherin, which is normally expressed in migrating neurons and mesenchymal cells during organogenesis, is upregulated in many invasive carcinoma cells, replacing the previously expressed E-cadherin.104
Research into the capability for invasion and metastasis has accelerated dramatically over the past decade as powerful new research tools, and refined experimental models have become available. Although still an emerging field replete with major unanswered questions, significant progress has been made in delineating important features of this complex hallmark capability. An admittedly incomplete representation of these advances is highlighted as follows.
The Epithelial-to-Mesenchymal Transition Program Broadly Regulates Invasion and Metastasis
A developmental regulatory program, termed the EMT, has become implicated as a prominent means by which neoplastic epithelial cells can acquire the abilities to invade, resist apoptosis, and disseminate.105,106,107,108,109,110 By co-opting a process involved in various steps of embryonic morphogenesis and wound healing, carcinoma cells can concomitantly acquire multiple attributes that enable invasion and metastasis. This multifaceted EMT program can be activated transiently or stably, and to differing degrees, by carcinoma cells during the course of invasion and metastasis.
A set of pleiotropically acting transcriptional factors (TF), including Snail, Slug, Twist, and Zeb1/2, orchestrate the EMT and related migratory processes during embryogenesis; most were initially identified by developmental genetics. These transcriptional regulators are expressed in various combinations in a number of malignant tumor types. Some of these EMT-TFs have been shown in experimental models of carcinoma formation to be causally important for programming invasion; others have been found to elicit metastasis when experimentally expressed in primary tumor cells.105,111,112,113,114 Included among the cell-biologic traits evoked by these EMT-TFs are loss of adherens junctions and associated conversion from a polygonal/epithelial to a spindly/fibroblastic morphology, concomitant with expression of secreted matrix-degrading enzymes, increased motility, and heightened resistance to apoptosis, which are implicated in the processes of invasion and metastasis. Several of these transcription factors can directly repress E-cadherin gene expression, thereby releasing neoplastic epithelial cells from this key suppressor of motility and invasiveness.115
The available data suggest that EMT-TFs regulate one another as well as overlapping sets of target genes. Results from developmental genetics indicate that contextual signals received from neighboring cells in the embryo are involved in triggering expression of these transcription factors in cells that are destined to pass through an EMT111; in an analogous fashion, heterotypic interactions of cancer cells with adjacent tumor-associated stromal cells have been shown to induce expression of the malignant cell phenotypes that are known to be choreographed by one or more of these EMT-TFs.116,117 Moreover, cancer cells at the invasive margins of certain carcinomas can be seen to have undergone an EMT, suggesting that these cancer cells are subject to microenvironmental stimuli distinct from those received by cancer cells located in the cores of these lesions.118 Although the evidence is still incomplete, it would appear that EMT-TFs are able to orchestrate most steps of the invasion-metastasis cascade, except perhaps the final step of colonization, which involves adaptation of cells originating in one tissue to the microenvironment of a foreign, potentially inhospitable tissue.
We still know rather little about the various manifestations and temporal stability of the mesenchymal state produced by an EMT. Indeed, it seems increasingly likely that many human carcinoma cells only experience a partial EMT, in which they acquire mesenchymal markers while retaining many preexisting epithelial ones. Although the expression of EMT-TFs has been observed in certain nonepithelial tumor types, such as sarcomas and neuroectodermal tumors, their roles in programming malignant traits in these tumors are presently poorly documented. Additionally, it remains to be determined whether aggressive carcinoma cells invariably acquire their malignant capabilities through activation of components of the EMT program, or whether alternative regulatory programs can also enable expression of these traits.
Heterotypic Contributions of Stromal Cells to Invasion and Metastasis
As mentioned previously, cross-talk between cancer cells and cell types of the neoplastic stroma is involved in the acquired capabilities of invasiveness and metastasis.94,119,120,121 For example, mesenchymal stem cells (MSC) present in the tumor stroma have been found to secrete CCL5/RANTES in response to signals released by cancer cells; CCL5 then acts reciprocally on the cancer cells to stimulate invasive behavior.122 In other work, carcinoma cells secreting IL-1 have been shown to induce MSCs to synthesize a spectrum of other cytokines that proceed thereafter to promote activation of the EMT program in the carcinoma cells; these
effectors include IL-6, IL-8, growth-regulated oncogene alpha (GRO-α), and prostaglandin E2.123
effectors include IL-6, IL-8, growth-regulated oncogene alpha (GRO-α), and prostaglandin E2.123
Macrophages at the tumor periphery can foster local invasion by supplying matrix-degrading enzymes such as metalloproteinases and cysteine cathepsin proteases76,120,124,125; in one model system, the invasion-promoting macrophages are activated by IL-4 produced by the cancer cells.126 And in an experimental model of metastatic breast cancer, tumor-associated macrophages (TAM) supply epidermal growth factor (EGF) to breast cancer cells, while the cancer cells reciprocally stimulate the macrophages with colony stimulating factor 1 (CSF-1). Their concerted interactions facilitate intravasation into the circulatory system and metastatic dissemination of the cancer cells.94,127
Observations like these indicate that the phenotypes of high-grade malignancy do not arise in a strictly cell-autonomous manner, and that their manifestation cannot be understood solely through analyses of signaling occurring within tumor cells. One important implication of the EMT model, still untested, is that the ability of carcinoma cells in primary tumors to negotiate most of the steps of the invasion-metastasis cascade may be acquired in certain tumors without the requirement that these cells undergo additional mutations beyond those that were needed for primary tumor formation.
Plasticity in the Invasive Growth Program
The role of contextual signals in inducing an invasive growth capability (often via an EMT) implies the possibility of reversibility, in that cancer cells that have disseminated from a primary tumor to more distant tissue sites may no longer benefit from the activated stroma and the EMT-inducing signals that they experienced while residing in the primary tumor. In the absence of ongoing exposure to these signals, carcinoma cells may revert in their new tissue environment to a noninvasive state. Thus, carcinoma cells that underwent an EMT during initial invasion and metastatic dissemination may reverse this metamorphosis, doing so via a mesenchymal-to-epithelial transition (MET). This plasticity may result in the formation of new tumor colonies of carcinoma cells exhibiting an organization and histopathology similar to those created by carcinoma cells in the primary tumor that never experienced an EMT.128
Distinct Forms of Invasion May Underlie Different Cancer Types
The EMT program regulates a particular type of invasiveness that has been termed mesenchymal. In addition, two other distinct modes of invasion have been identified and implicated in cancer cell invasion.129,130 Collective invasion involves phalanxes of cancer cells advancing en masse into adjacent tissues and is characteristic of, for example, squamous cell carcinomas. Interestingly, such cancers are rarely metastatic, suggesting that this form of invasion lacks certain functional attributes that facilitate metastasis. Less clear is the prevalence of an amoeboid form of invasion,131,132 in which individual cancer cells show morphologic plasticity, enabling them to slither through existing interstices in the ECM rather than clearing a path for themselves, as occurs in both the mesenchymal and collective forms of invasion. It is presently unresolved whether cancer cells participating in the collective and amoeboid forms of invasion employ components of the EMT program, or whether entirely different cell-biologic programs are responsible for choreographing these alternative invasion programs.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


