Gene/protein
Function
Alteration
Type 1 (%)
Type 2 (%)
MSI
OG
Hypermethylation
20–45
11
ERBB2 (HER2)
OG
Amplification
10–30
18–80
PTEN
TSG
Inactivation
83
11
PIK3CA
OG
Mutation
26–36
5
PIK3R1
TSG
Mutation
21
6
ESR1/PGR (ER/PR)
TF
Loss of expression
30
81
KRAS
OG
Mutation
10–30
10
FGFR2
OG
Mutation
10
3
CTNNB1
OG
Mutation
14–44
5
CDH1
TSG
Hypermethylation
10–20
60–90
TP53
TSG
Mutation
10–20
90
ARID1A
TSG
Mutation
19
26 (in CCC)
CDKN2A (P16)
TSG
inactivation
10
40–45
PPP2R1A
TSG
Mutation
5
32
STMN1
OP
Overexpression
15
64
GPER
TF
Loss of expression
23
63
Gene | Low-grade EEC (%) | High-grade EEC (%) | Serous carcinoma (%) | Carcinosarcoma (%) |
|---|---|---|---|---|
PTEN | 67 | 90 | 2.7 | 33.3 |
PIK3CA | 38 | 56.7 | 27 | 28.6 |
ARID1A | 46.7 | 60 | 10.8 | 23.8 |
KRAS | 16.6 | 26.7 | 8.1 | 16.7 |
CTNNB1 | 23.8 | 20 | 2.7 | 4.8 |
TP53 | 10.1 | 30 | 67.6 | 64.3 |
PPP2R1A | 6.9 | 10 | 43.2 | 21.4 |
Table 15.3
Molecular testing of endometrial tumors
Protein name | Gene | Normal gene/protein function | Test methodology/specimen | Clinical utility/clinical application |
|---|---|---|---|---|
MSI (MLH1, MSH2, MSH6, PMS2) | MLH1, MSH2, MSH6, pMS2 | DNA replication | IHC, MLH1 promoter methylation, MSI testing by PCR and DNA sequencing/paraffin-embedded tissue | Screening of endometrial carcinomas |
ERBB2 (HER2) | ERBB2 | Multifunctional; activation of downstream pathways | FISH, CISH or IHC/paraffin-embedded tissue | Potential predictor of response to specific agents |
PTEN | PTEN | tumor suppressor/protein tyrosine phosphatase | Single-strand conformation polymorphism, DNA sequencing and IHC/paraffin-embedded tissue | Mutation associated with low-grade and low-stage with improved survival |
PI3Kα | PIK3CA | Oncogene | PCR-based sequencing of DNA/paraffin-embedded tissue | |
Fusion protein | JAZF1-SUZ12 t(7;17) | JAZF1: transcriptional repressor; SUZ12: zinc finger domain | Chromosomal karyotyping, RT-PCR on RNA isolated from paraffin-embedded tissue and FISH | Diagnosis of a subset of low grade endometrial stromal sarcomas |
Fusion protein | YWHAE-NUTM2A or YWHAE-NUTM2B t(10;17) | Ubiquitously expressed regulator proteins | RT-PCR and FISH/paraffin-embedded tissue | Diagnosis of a subset of high grade endometrial stromal sarcomas |
Microsatellite Instability (MSI)
Hereditary non-polyposis colorectal cancer (HNPCC; Lynch syndrome (LS)) is an autosomal dominant disorder that is attributable to a germline mutation in one or more of the 4 DNA mismatch repair (MMR) genes: MSH2 and MSH6 (chromosome 2), MLH1 (chromosome 3), and PMS2 (chromosome 7) [8]. The genes may be inactivated, however, by other mechanisms, including somatic MLH1 promoter methylation, miRNA overexpression, or rarely, epigenetic silencing of MSH2 through deletion of the adjacent EPCAM gene.
Dysfunction or loss of the MMR system eventuates in DNA replication errors to the genome, some of which are seen as alterations in the lengths of short repeated sequences known as microsatellites, and some of which eventuate in the development of cancers. Patients with LS are at increased risk for cancers from numerous sites, including a 40–60 % lifetime risk of both endometrial and colorectal cancers, and a 9–12 % lifetime risk of ovarian cancer [9].
Although only 2–5 % of endometrial carcinomas are LS-related, 50–60 % of LS patients will present first with an endometrial carcinoma primary [10–12]. Most endometrial carcinomas associated with the LS have alterations in MSH6, MSH2 or MLH1. The diagnosis of a LS-associated endometrial carcinoma not only allows the possibility of counseling and increased surveillance for the index patient’s relatives who are at risk, but may also allow the earlier detection of second LS-associated malignancy in the index patient [13, 14]. Additionally, an LS-associated carcinoma may have prognostic significance for the index patient, as there is some preliminary evidence that they tend to be more aggressive, although the available data is not entirely congruent on this point [10, 15, 16].
There is currently an ongoing debate about the level of testing of endometrial carcinomas that is required to maximize the identification of the 2–5 % that may be associated with LS. Tumor-associated characteristics have been proposed to identify cases in need of further testing, since MMR-abnormal tumors (with the possible exception of MSH6-altered tumors) tend to show frequent tumor infiltrating lymphocytes, undifferentiated or dedifferentiated areas, and lower uterine segment origin [17, 18]. Other features of LS-associated pathology include type II endometrial carcinomas in younger patients, and an overrepresentation of the endometriosis-associated tumors namely; endometrioid and clear cell carcinoma (CCC) in the ovary [19].
Traditional Amsterdam I and II, Society of Gynecologic Oncologists (SGO), and revised Bethesda criteria are now recognized to be insensitive for identifying older LS patients and morphology-based criteria are also similarly insensitive as a primary strategy [20–26]. The SGO, reflecting a progressive change in sentiment toward a more universal screening approach, has recently endorsed a more expansive strategy, endorsing “A more sensitive strategy” to detect LS that includes “universal molecular tumor testing for either all endometrial cancers or cancers diagnosed at age less than 60 years, regardless of personal or family cancer history” if resources are available [27]. The Stanford group recently noted that “41 % with an LS-associated germline mutation were not associated with any of the traditional indicators that have been recommended for LS screening (i.e., age 50 years or younger, personal/family cancer pedigree that meets Bethesda guideline criteria, presence of MMR-associated tumor morphology, or location in the lower uterine segment)” [28]. The authors recommended universal testing for all newly diagnosed endometrial carcinomas [28]. A long-standing group of European experts recently recommended that all women diagnosed with endometrial cancer up to the age of 70 years be offered tumor testing by MSI or IHC [29]. Others have endorsed a more hybrid strategy that combines universal testing by MSH6 IHC and selective testing of MLH1, PMS2, and MSH2 IHC on the basis of clinicopathologic parameters [30].
A consensus seems to be emerging around universal testing for all histotypes up to the age of 60 years. This approach has been found to display the best performance in two recent studies, and entailed the testing of all endometrial carcinomas by MMR IHC in patients younger than 60 years, tumor MLH1 methylation in patients with MLH1 IHC loss, and germline mutations in patients exhibiting loss of MSH6, MSH2, or PMS2 or loss of MLH1/PMS2 with absence of MLH1 methylation [20, 31].
Testing Strategy
Overall, MSI testing, IHC, methylation analyses, MMR gene sequencing, and EPCAM gene analyses are tests that in total may be applied to diagnose LS-related endometrial carcinomas.
MSI Testing
Testing for MSI is performed by a comparative analysis of the polymerase chain reaction (PCR)-amplified microsatellite fragments between the tumor and normal tissue from the same patient. Paraffin-embedded tissue (tumor) and normal tissue (blood or paraffin-embedded tissue) are used for the test [32]. A panel of five microsatellite markers referred to as the Bethesda panel has been recommended for initial screening. This panel includes two mononucleotide (BAT-25 and BAT-26) and three dinucleotide (D5S346, D2S123, and D17S250) repeats. Samples with instability in at least two of these markers are defined as MSI-High (MSI-H), whereas those with one unstable marker are designated as MSI-Low (MSI-L) and samples with no detectable alterations are microsatellite-stable (MSS) [33]. Testing a secondary panel of mononucleotide markers, such as BAT-40, to exclude MSI-L in cases in which only the dinucleotide repeats are mutated has been recommended [23] (Fig. 15.1). Approximately 20–23 % of endometrial carcinomas show high frequency MSI-H, and approximately 80 % of these are due to an epigenetic inactivation (methylation) of the MLH1 promoter [20]. MSI testing would be ineffective as a primary screening modality without additional testing because (1) approximately 10–30 % of sporadic EECs are MSI-H [34], and less than 20 % of these are true LS-associated cancers [35] and (2) A significant subset of MSH6 mutated endometrial carcinomas are MSS [36, 37].
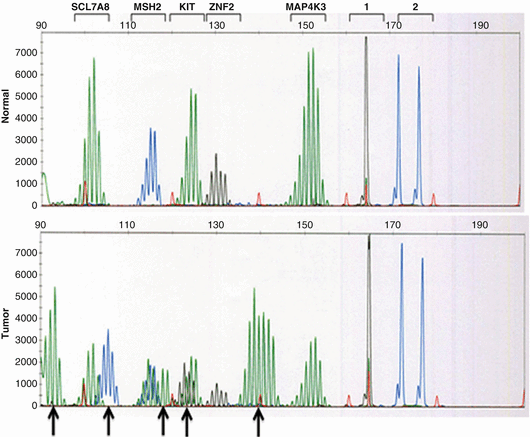
Fig. 15.1
Electropherograms illustrating MSI in tumor DNA. Patient DNA is extracted from both tumor and adjacent normal tissue and amplified using a multiplex PCR kit that includes oligonucleotide primer pairs for 5 mononucleotide markers and 2 pentanucleotide markers. Comparison of the two patterns indicates microsatellite instability at 5 of 5 mononucleotide markers as indicated by the arrows. Pentanucleotide markers are included for identity to ensure that comparison testing is performed on tissue obtained from the same individual (Courtesy Professor Cindy Vnencak-Jones, Vanderbilt University)
Immunohistochemistry
IHC is a reasonably sensitive and specific approach for identifying the specific MMR that is altered in endometrial carcinoma, and for screening all endometrial carcinoma under a universal strategy [11, 38]. In one analysis, IHC with MLH1 and MSH2 alone detected 69 % of MSI-H tumors with a specificity of 100 %. The inclusion of the other two markers (PMS2 and MSH6) to the panel increased the sensitivity to 91 %, but decreased the specificity to 83 %. MSH6 loss by IHC primarily was responsible for the decreased specificity, as it did not correlate as well as the others with MSI [36]. MSH2 and MSH6 are obligate dimerization partners, as are MLH1 and PMS2. Each member of the pair is generally unstable when unpaired. As such, a defect in one gene that eventuates in loss of expression of its corresponding protein is typically accompanied by loss of expression of its partner [39]. Interpretation of MMR IHC should only be performed in conjunction with internal controls with retained expression [40]. The latter may be stroma, endothelial cells, inflammatory cells, or myometrium. Additionally, care must be taken not to interpret foci of atypical hyperplasia, which are frequently admixed with, or adjacent to endometrial carcinoma. Finally, MMR protein expression may be artifactually altered by the lack of prompt or adequate formalin fixation, and this possibility should be considered especially in cases with only focal or equivocal expression. Parenthetically, MSI has also been seen in approximately 20 % of endometrial hyperplasia, suggesting that MSI may be an early event in the genesis of EEC [41].
MMR Gene Sequencing
In a recent analysis of minimally selected endometrial carcinoma patients, 5.9 % (7/118) showed a germline mutation: 4 had MLH1, 2 had MSH6, and 1 had MSH2 [20]. In another study, germline mutation were identified in 3 % [31]. The current approach includes direct gene sequencing on DNA extracted from fresh tissue or peripheral blood, to detect genomic insertion/deletion rearrangements of the MMR genes and/or deletions in the 3′ end of EPCAM where they most commonly occur [31].
DNA Methylation Analysis
As previously noted, the vast majority of endometrial carcinoma with abnormal MMR protein expression has loss of MLH1 due to methylation of the corresponding MLH1 promoter. As such methylation analysis is generally performed on tumors showing MLH1/PMS2 protein loss. A common approach to methylation analyses is the use of sodium bisulfite conversion and quantitative MethyLight assays [42]. MLH1 methylation is generally quantitatively reported based on the percentage of methylated reference (PMR) calculations, using ≥ 4 % PMR as cutoff for positive methylation status [31]. BRAF (V600E) mutational and/or IHC analyses, which are useful in distinguishing sporadic, MSI-H colorectal cancers from LS-associated MSI-H cancers, are of less utility in endometrial carcinomas, since BRAF mutations are extraordinarily infrequent in endometrial carcinomas [43–45].
ERBB2 (HER2)
Erb-b2 receptor tyrosine kinase 2 (ERBB2 also known as HER2), is a member of the human epidermal growth factor receptor (EGFR) family of tyrosine kinases. A ligand binding of these transmembrane receptors result in their dimerization and intracellular domain phosphorylation, with eventual activation of pathways associated with proliferation and differentiation. In general, ERBB2 (HER2) alterations are significantly more frequently found in non-endometrioid (NEEC) as compared with EECs [46]. ERBB2 alterations have been most extensively studied in ESC, where ERBB2 overexpression as assessed by immunohistochemistry (IHC) has reportedly ranged from 14 % to 80 %, and ERBB2 gene amplification as assessed by fluorescent in situ hybridization (FISH) has reportedly ranged from 21 % to 47 % [47–63]. The significance of ERBB2 overexpression and/or amplification (alterations) is controversial. Regarding prognosis, most studies have reported an association between ERBB2 alterations and poor survival [50, 54, 55, 62] but other studies have found no such association [56, 64]. The role of targeting ERBB2 protein has also been assessed in isolated reports [47, 65, 66]. However, to date, prospective trials have failed to show any significant therapeutic effect to this approach in patients with ESC. A randomized phase II study (ClinicalTrials.gov Identifier: NCT01367002) is currently being conducted in patients with advanced stage or recurrent 3+ ERBB2 overexpressing or FISH-amplified ESC, assessing carboplatin/paclitaxel with or without the anti-ERBB2 (HER2) agent trastuzumab. Nonetheless, in vitro studies continue to demonstrate the effectiveness of therapeutic approaches targeting the ERBB2 protein in serous carcinoma cell lines, including T-DM1, an antibody–drug conjugate, and neratinib, an ERBB1 and ERBB2 inhibitor [67, 68]. Taselisib, a selective inhibitor of PIK3CA, has been shown to be effective on PIK3CA-mutated and ERBB2 amplified ESC in vitro and in vivo, whereas GDC-0980, an inhibitor of class I PI3 kinase and mTOR kinase (TORC1/2), and AZD8055, a novel dual mTORC1/2 inhibitor have similarly been shown to be effective in ESC cell lines showing oncogenic PIK3CA mutations and ERBB2 gene amplification in vitro [69–71].
There are currently no well-established guidelines for ERBB2 testing in endometrial carcinoma. The staining pattern by IHC is often basolateral or lateral, without the circumferential staining that is characteristic of ERBB2-amplified breast cancers, and the staining is often heterogeneous within a given tumor [72, 73]. The latter suggests that large tumor sections are preferable for this testing, and that results from small samples, such as biopsies and curettages be viewed with some skepticism. Direct extrapolation of breast scoring criteria to the endometrium probably results in a significant overestimation of the rate of ERBB2 amplification when IHC is used as the primary means of its estimation [73]. One recent study found that the overall concordance rate between chromogenic in situ hybridization (CISH) and IHC was 64 % with the 2007 ASCO/CAP criteria [74] and 32 % with the 2013 ASCO/CAP criteria for breast cancers [75]. Another study, using FISH and IHC and the 2007 ASCO/CAP criteria [74] found the concordance rate to be approximately 75 % [72]. Therefore, the breast 2007 ASCO/CAP scoring criteria [74] appears to be more applicable to ESC. Ultimately, responsiveness to therapy is the gold standard for assessing the effectiveness of these modalities for assessing ERBB2 alterations, and future studies will be required to determine the criteria that show the strongest correlation with therapeutic efficacy.
Phosphatase and Tensin Homolog (PTEN)
PTEN is a tumor suppressor gene located on 10q23 that encodes a dual-specificity phosphatase. The primary substrate of the PTEN protein is the phospholipid phosphatidylinositol-[3–5]-trisphosphate (PIP3) [76]. Production of PIP3 leads to translocation and activation of the protein kinase AKT which mediates cell survival and proliferation. PTEN acts as the negative check-point on the level of PIP3 to down regulate cell proliferation [77]. Germline mutations in the PTEN gene have been identified in Cowden Syndrome and Lhermitte-Duclos disease [78, 79]. On the other hand, somatic mutations have been demonstrated in a wide variety of tumors, including prostate cancer [80] and melanoma [81]. In addition to gene mutation, PTEN can be inactivated by deletion, epigenetic silencing, disruption of competitive endogenous RNA networks, micro-RNA regulation, posttranslational modifications, and aberrant localization [82]. PTEN mutations appear to be early events in type I endometrial carcinogenesis, being present in 50 % of complex atypical hyperplasia [83]. In endometrial carcinomas, PTEN gene mutations are more frequently identified in EECs (80 %) than non-EECs, and accordingly may have prognostic significance [84–87].
PTEN mutations have been identified in ~80 % of EECs with MSI-H compared to ~30 % of tumors that are MSI-stable [87]. It appears that replication error may predispose tumors to develop mutations in the PTEN gene even before clinicopathologic detection of cancer or precancerous lesion [88]. It has been reported that cells with PTEN inactivation in EEC are sensitive to mTOR inhibitors such as everolimus and temsirolimus [89, 90]. In a phase II study in patients with advanced or recurrent endometrial cancer, 26 % patients had a partial response and 63 % had stable disease on temsirolimus [89, 91].
Mutational and IHC Analyses
Substitution missense, deletion frameshift, substitution nonsense, and insertion frameshift are the most common mutations reported in the PTEN gene in EEC in descending order of frequency [92]. Single-strand conformation polymorphism (SSCP) analysis and DNA sequencing are the main molecular testing techniques that have been applied to study PTEN mutations [93]. The latter have been reported in exons 8, 7, 5, 6 3, 1, and exon 2. No mutations reported in exons 4 and 9 [93]. PTEN status by IHC correlates with the mutational status of the gene with statistically significant lower staining in PTEN mutated tumors than in tumors without alterations [94]. However, PTEN IHC is acutely affected by variety prenanalytic variables, and accordingly must be carefully validated and constantly monitored [94].
The PTEN protein shows nuclear and cytoplasmic distribution with loss/weak expression in cases with molecular alteration compared to high expression in cases without mutation, and scoring is most correlated with gene status if both intensity and extent of staining are considered [94] An inverse relationship between the level of PTEN protein and phosphorylated AKT protein using IHC has been identified [95].
Phosphatidylinositol 3-Kinase, Catalytic, Alpha (PIK3CA) and Regulatory Subunit 1 (PIK3R1)
PI3Kα (PI3K-alpha) is a protein complex composed of a catalytic, p110α, subunit and regulatory, p85α, subunit encoded by the PIK3CA (3q26.32) and PIK3R1 (5q13.1) genes, respectively [96]. Mutation of PIK3CA results in increase in AKT-dependent or independent signaling and leads to increased cell proliferation, growth, survival, and migration [97]. This pathway is antagonized by PTEN phosphatase [98]. One of the common mechanisms of PI3K-alpha activation is the somatic gain-of-function mutations within PIK3CA, PIK3R1 or loss of the PTEN activity [98, 99]. The product of the PIK3R1 gene, binds to p110α leading to stabilization and inhibition of p110α. Somatic mutations in PIK3R1 have been reported in few tumor types compared to that of PIK3CA [99, 100]. Somatic mutation in PIK3R1 has been reported in 43 % of EECs and 12 % of NEECs especially PIK3CA-wild type EECs than in PIK3CA mutant ones [99].
Somatic PIK3CA mutations have been reported in 52 % of EECs and 33 % of NEECs [92, 101, 102]. Fifty percent of all nonsynonymous PIK3CA mutations were localized in exons 1–7 and the other half were in exons 9 and 20. Approximately 62 % of exons 1–7 mutants and 64 % of exons 9–20 mutants were activating [92, 98, 101, 103].
Hotspots at amino acids 88, 93, and 111 within the ABD, C2 domain and the linker constitute the most common mutations affecting exon 1–7. Mutational analysis by nucleotide sequencing of all coding exons (exons 1–20) of the PIK3CA gene has been reported [92].
Endometrial Stromal Sarcoma and Undifferentiated Endometrial Sarcoma
Low grade endometrial stromal sarcomas (LGESS) are genetically heterogenous, but significant subsets are driven by recurrent genetic alterations. Translocation t(7;17)(p15;q21), leading to the fusion of two zinc finger genes, JAZF1 (juxtaposed with another zinc finger gene 1) and JJAZ1 (SUZ12) (joined to JAZF1; suppressor of zeste-12 protein, also known as SUZ12 polycomb repressive complex 2 subunit) is a distinctive molecular alteration identified in 23–80 % of LGESS [104–106]. The sensitivity of the JAZF1-SUZ12 fusion appears to be highest in LGESS with classic morphology [107]. A subgroup of LGESS has been found to have translocations involving 6p21 [104, 108]. PHF1 (PHD finger protein 1) gene in 6p21 was reported to be recombined with JAZF1 gene showing a 6p;7p rearrangement, which results in the formation of a JAZF1-PHF1 fusion gene and with EPC1 (enhancer of polycomb) gene in 10p11 that had a 6;10;10 translocation [109]. Additionally, rare endometrial stromal sarcoma cases were reported with a t(X;17)(p11.2;q23) and a t(10;17)(q22;p13) [110, 111]. Other reported chimeric fusions include in LGESS include the MEAF6-PHF1, ZC3H7-BCOR, and the recently described MBTD1–CXorf67 [112, 113]. Most studies reported an absence of these characteristic alterations in high grade endometrial stromal sarcomas (HGESS) and undifferentiated endometrial sarcomas [108]. Cytogenetic chromosomal karyotyping [114], reverse transcriptase-PCR (RT-PCR) with RNA isolated from paraffin-embedded tissue samples [104, 105] and FISH on paraffin-embedded tissue for the detection of the fusion protein [104] are the different tools to test for t(7;17).
Recently, a subset of HGESS have been defined by the presence of t(10;17)(q23;p13), which results in the genetic fusion between the YWHAE gene and 1 of 2 nearly identical NUT family members: NUTM2A or NUTM2B [115–118]. This subset is more clinically aggressive than their JAZF1:SUZ12-harboring low grade counterparts, but it is unclear whether they bear clinical significance relative to other HGESS. Nonetheless, these tumors need to be segregated. RT-PCR and FISH have both been applied to test for these alterations, and there is a 94 % concordance between these modalities [117]. It has recently been reported that diffuse immunoreactivity for cyclin D1 (CCND1), in the appropriate morphologic context, is an excellent surrogate marker for YWHAE-altered endometrial stromal neoplasms [118].
Molecular Testing of Ovarian Cancer
Epithelial ovarian cancer is the most common cause of cancer related deaths among gynecological cancers in the western countries with an estimated 21,980 new cases and 14,270 deaths in 2014 [120]. This high mortality is probably because most cases of ovarian cancers present at an advanced stage. The current practice consists of surgical excision of tumors, followed by platinum–taxane based chemotherapy [121]. Unfortunately, despite aggressive treatment, most cancers recur with dismal 5-year survival rate at ~ 45 % [122]. Molecular alterations in ovarian cancer have been the subject of research with the goal of identifying diagnostic, predictive, prognostic, and/or therapeutic targets. Up to 20 % of high grade ovarian cancers are associated with germline mutations in BRCA1 or BRCA2 [123]. On the other hand, somatic alterations in BRCA1/2 are seen in approximately 50 % in ovarian cancers [124].
Morphologically, ovarian epithelial tumors are classified into two groups with distinct molecular profiles (Table 15.4). Type I tumors include low grade serous carcinomas (LGSC), clear cell carcinoma (CCC), low and intermediate grade ovarian endometrioid carcinoma (OEC), and mucinous carcinoma. These tumors usually present at earlier stages, grow slowly and confined to the one or both ovaries. BRAF and KRAS somatic mutations are common molecular alterations in these tumors. Type II ovarian tumors include high grade ovarian serous carcinoma (HGOSC), high grade OEC, carcinosarcoma and poorly differentiated carcinomas. These tumors are aggressive with high metastatic potential at presentation. Approximately 80 % of HGSC exhibit TP53 mutation [124–126]. PIK3CA and RAS are other signaling pathways which have been found to be altered in 45 % of HGOSC [124]. Table 15.5 summarizes the most common molecular testing in ovarian tumors.
Mutation | Overall | HGSC | LGSC | Clear Cell | EC | Mucinous |
|---|---|---|---|---|---|---|
BRAF | 11 % [125] | <1 % [124] | 24–33 % [181] | 1 % [182] | 24 % [183] | 50–75 % [184] |
KRAS | 11 % [125] | <1 % [124] | <1 % [183] | 50–75 % [184] | ||
PIK3CA | <1 % [124] | 5 % [181] | 20 % [185] | Rare | ||
PTEN | 20 % [125] | <1 % [124] | 20 % [126] | Rare |
Table 15.5
Molecular testing of ovarian tumors
Protein name | Gene | Normal gene/protein function | Test methodology/specimen | Clinical utility/clinical application |
|---|---|---|---|---|
BRCA | BRCA1&2 | Repair of damaged to DNA | DNA sequencing of the coding regions on blood sample IHC/paraffin-embedded tissue | Genetic counseling and testing of family members |
P53 | TP53 | Tumor suppressor | PCR based sequence analysis and IHC/paraffin-embedded tissue | High grade serous carcinomas harbor p53 mutation |
BAF250(ARID1A) | ARID1A | Tumor suppressor | Sequence analysis of the entire coding region and IHC/paraffin-embedded tissue | Most common on clear cell carcinomas |
FOXL2 | FOXL2 | Transcription factor | PCR and Pyro-sequencing/paraffin-embedded tissue | Specific for adult granulose cell tumors |
BRCA1 and BRCA2
Hereditary ovarian cancers represent approximately 5–12 % of invasive ovarian carcinomas, with mutations in the BRCA1 and BRCA2 genes accounting for the majority of these cases [127]. The lifetime risk of ovarian carcinoma in carriers of germline mutation is approximately 20–40 % with an earlier onset with BRCA1 mutations [128]. Identification of families with germline mutations of the BRCA1 and BRCA2 genes is important for genetic counseling and testing of these families [123].
BRCA1 gene is involved in cell growth, cell cycle checkpoint control and repair of damage to DNA by forming several complexes through association with different adaptor proteins [129]. BRCA1 is a tumor suppressor gene that regulates other genes including MYC and CCND1 (cyclin D1) [130, 131]. Subjects with hereditary predisposition usually inherit one defective copy and the other copy of BRCA1 will develop somatic inactivating mutation with subsequent elimination of the protein expression in manifest malignant cells.
Twenty percent of HGOSC displayed germline or somatic mutations in BRCA1 or 2 in the TCGA analysis [124]. In that same analysis, 11 % had lost BRCA1 expression through DNA hypermethylation; epigenetic silencing of BRCA1 was found to be mutually exclusive of BRCA1/2 mutations [124].
The BRCA1 gene has 24 exons with exon 11 being the longest that encodes ~60 % of the protein (220 KDa). Mutations have been described among Ashkenazi Jewish patients with breast and ovarian cancers with 185delAG and 5382insC being the most common founder mutations. Most mutations result in protein truncation or loss of function of the BRCA domain that regulates gene transcription [130, 131]. In addition to germline mutation, BRCA1 can be lost by a variety of other mechanisms including somatic mutation and promoter hypermethylation [124].
BRCA2 gene contains 27 exons encoding a 384 kDa nuclear protein. Exon 10 and 11 being the largest; the latter encodes a protein hat binds RAD51 protein that involves in DNA repair. This indicates that BRCA2 is also involved in DNA repair like that of BRCA1. Among the Ashkenazi Jewish population, 6174delT is found in 1.5 % [132]. Inactivation of the BRCA1/2 genes by hypermethylation of the promoter region occurs frequently in 40–50 % of sporadic HGOSCs compared to the germline mutation in hereditary cases [133].
BRCA mutations and/or methylations not only have prognostic significance but may be predictive of responsiveness to PARP inhibitors [134–136]. TCGA data suggests that there is better overall survival for BRCA1/2 mutated cases of HGOSC than BRCA1/2 wild-type cases [124]. For BRCA1, the improved prognosis is present even for epigenetically silenced cases relative to their wild-type counterparts. Although it has been less robustly studied, low levels of BRCA1 protein, as assessed by IHC, may also be a favorable prognostic factor in uterine serous carcinomas [137].
Screening and Testing for BRCA1/2 Mutations
The National Comprehensive Cancer Network has established guidelines for screening individuals and families with breast/ovarian cancer. The criteria are (1) A personal history of breast cancer diagnosed at or before the age of 45 years, bilateral breast cancer, or ovarian cancers (2) A personal history of ovarian cancer at or before the age of 50 years with a close relative (parent, sibling, child, grandparent, grandchild, uncle, aunt, nephew, niece or first cousin) diagnosed with ovarian or breast cancer (3) A history of breast cancer at a young age in at least two close relatives (4) A male relative with breast cancer (5) A family member who has both breast and ovarian cancers or bilateral breast cancer (6) Two or more close relatives with ovarian cancer (7) Known BRCA1/2 kindred, (8) Ashkenazi Jewish ancestry, with a personal history of ovarian cancer at any age or has a close relative with breast or ovarian cancer.
The mutation analysis is performed on a blood sample. In populations with known mutations, e.g., Ashkenazi Jews, initial screening for 185delAG and 5382insC (BRCA1) and 6174delT (BRCA2) is offered. However, in the general population with suspected BRCA mutation, comprehensive sequencing of the coding regions and exon/intron boundaries are offered. Even sequencing may fail to detect all mutations, including large deletions on one allele and some splice mutations [138]. BRCA testing as part of multi-gene panel template is also available. A 6-gene (BRCA1, BRCA2, TP53, PTEN, STK11, and CDH1) panel as a first approach test in patients who are candidates for BRCA1/2 testing has been reported, with a reported advantage of identifying mutations in less common susceptibility genes [139].
In recent studies, BRCA1 IHC has been identified to be a highly reproducible and accurate surrogate for detecting germline, somatic, and epigenetic mechanisms of BRCA1 loss in HGOSC. In one such report, BRCA1 IHC had a high negative predictive value (NPV) of 95 % but a low positive predictive value (PPV) of 52 % in correlation with the germline BRCA1 status. In correlation with promoter hypermethylation and somatic mutations of BRCA1 loss, the PPV was 88 % [140, 141]. The authors scored the IHC results for positive nuclear staining as loss (<5 % positive), equivocal (5–10 % stain “less intense” compared with the internal control), and retained (>10 % positive or >5 % when staining intensity of tumor cell nuclei is similar to the internal control) [140, 141].
Morphologic features of ovarian serous carcinomas may also be predictive of BRCA alterations. Soslow et al. [142] compared BRCA1 altered cases (germline mutated, promoter methylated, or somatic mutated) with controls, and found that BRCA1-associated tumors more frequently had the so-called “SET” morphologic features (Solid, pseudo-endometrioid, and transitional cell carcinoma-like morphology), tumor infiltrating lymphocytes, higher mitotic indexes, and necrosis (geographic or comedo). BRCA2-associated cases also showed “SET” morphologic features but did not display a prominent component of necrosis and tumor infiltrating lymphocytes [142].
Tumor Protein p53 (TP53)
TP53 (17p13.1), encoding the p53 (TP53) protein is the most common altered tumor suppressor gene in human neoplasms. P53 is a tetrameric transcription factor that regulates cell growth, death and DNA integrity by controlling several target genes [143]. TP53 mutations, tested by conventional mutational analysis or inferred from overexpression, or complete absence, of protein by IHC, are recognized in HGOSC. Sixty to seventy percent of HGOSC cases that have been tested by full TP53 (p53) gene sequencing harbor TP53 mutations [144]. In contrast, EOC, CCC, and mucinous carcinomas have a much lower incidence of TP53 mutations [145]. TP53 mutations have been reported in serous tubal intraepithelial carcinomas. This suggests that TP53 mutation is an early event in the development of HGOSC of the ovary, fallopian tube, and peritoneum [146]. Inactivation of the TP53 gene may be an important event for the initiation of serous cancers even in those cancers that appear to retain the wild type sequence or IHC staining. Other mechanisms which may lead to TP53 (p53) inactivation include promoter or splicing alterations. Inactivation of TP53 (p53) has been reported to be associated with resistance to cytotoxic therapy [147].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


