(1)
Institut Bergonié, Bordeaux, France
Abstract
Signalling pathways originating from the interaction between growth factors (GF) and tyrosine kinase receptors (TKRs) are certainly the best known because of their leading role in oncogenesis. These pathways are multiple and will be studied in several chapters; the present chapter exclusively concerns GFs and TKRs, whereas Chaps. 2 and 3 will present two major signalling pathways downstream these interactions. We adopt here a definition of ‘growth factors’ restricted to those activating TKRs. Other classes of receptors will be studied in other chapters: receptors activating cytoplasmic tyrosine kinases (Chap. 4), serine/threonine kinase receptors (Chap. 5), etc.
More than 100 GF and 58 TKRs, distributed among 20 families, have been identified and a large part of them may play a role in oncogenesis. Activation of TKRs occurs according to a unique mechanism, involving dimerisation and autophosphorylation of the receptors, through their own tyrosine kinase activity, and constitutes the starting point of the signalling pathway. These dimeric associations of TKRs establish the link between GFs and cellular response; there exists a complex combinatorial pattern of these associations, which determines the type of action exerted on the cell receiving the signal. The number of possible combinations is high, which explains why the same signal may generate distinct consequences in the target cells: for instance, proliferation and differentiation may be induced by the same signal acting on different cells. Lastly, the signalling pathways downstream the GF–TKR interaction are multiple and depend upon the proteins expressed by the target cell, which is often referred to as ‘the cellular context’.
1.1 General Features of the GF–TKR Interaction
1.1.1 Overview
The various GFs and TKRs can be grouped in families of homologous proteins: for instance, the GFs of the epidermal growth factor (EGF) family interact with the TKRs of the epidermal growth factor (EGFR) family, and so on. The denominations ‘epidermal’ or fibroblastic’ growth factors have more to do with the circumstances of their discovery than with the specificity of their target: epithelial tissues may express fibroblastic growth factors (FGF) and non-epithelial tissues may express epidermal growth factors.
Growth factors are polypeptides transmitting mitogenic messages, i.e. messages able to trigger mitosis. They are secreted by numerous cell types, not organised in individualised glands, and act mostly in the close vicinity of the cell of origin. By analogy (and opposition) to the endocrine system, comprised of hormones circulating in blood, growth factors are said to be paracrine factors. They may also act as juxtacrine factors (when the cell of origin and the target cell are linked by tight junctions) and as autocrine factors (when they directly act on the cell which secreted them). Whereas the cellular response to GFs, as mediated by TKRs, has been deciphered to a high degree of molecular understanding, as presented below, the regulation of the expression and secretion of GFs by the cells of origin has not been studied in depth, and we have no comparable knowledge of the pathways involved, which would be of utmost importance in oncogenesis.
Table 1.1 presents the 20 classes of TKRs and the corresponding GFs. All TKRs possess a single transmembrane domain; a homologous intracellular domain bearing the catalytic centre, of limited structural variety; and an extracellular part which is highly variable from one class to another. This extracellular part contains various protein domains characterised by specific amino acid sequences (cysteine-rich domains, acidic domains, immunoglobulin-like domains, etc.), which are presented on Fig. 1.1. The mechanism of activation of all TKRs is the same: after GF binding occurs a step of dimerisation, which triggers the autophosphorylation of tyrosine residues located in the intracellular domain, close to the C-terminal part of the receptor. The presence of phosphotyrosine residues constitutes the true signal that permits the implementation of a transduction pathway leading to the activation of intracellular effectors.
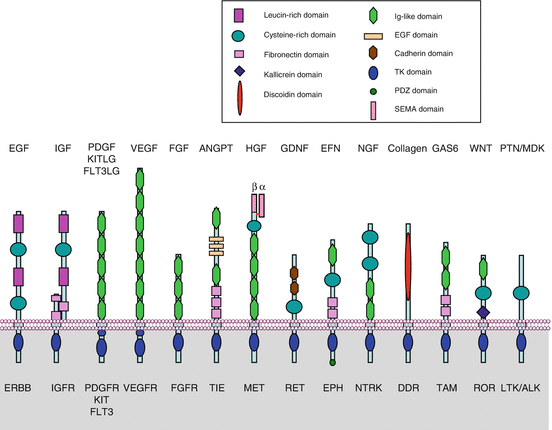
Table 1.1
Families of tyrosine kinase growth factor receptors
Class | Family | Receptors | Ligands |
|---|---|---|---|
Class I | EGF receptors | EGFR ERBB2 (no ligand) ERBB3 (no TK activity) ERBB4 | EGF TGFα HBEGF Epigen (EPGN) Epiregulin (EREG) Betacellulin (BTC) Amphiregulin (AREG) Neuregulins 1 to 4 (NRG1 to NRG4) |
Class II | Insulin receptors | INSR IGF1R [IGF2R]a INSRR | Insulin IGF1 IGF2 |
Class III | PDGF receptors and related receptors | PDGFRα, PDGFRβ KIT FLT3 CSF1R | PDGFA to PDGFD SCF (KITLG) FLT3LG CSF1, IL34 |
Class IV | FGF receptors | FGFR1 FGFR2 FGFR3 FGFR4 | FGF1 to FGF14 FGF16 to FGF23 |
Class V | VEGF receptors | FLT1 (VEGFR1) KDR (VEGFR2) FLT4 (VEGFR3) | VEGFA, VEGFB, PlGF VEGFA VEGFC, VEGFD |
Class VI | HGF receptors | MET MST1R (RON) | HGF (SF) MST1 |
Class VII | NTRK receptors | NTRK1 NTRK2 NTRK3 | NGF BDNF NT3, NT4 |
Class VIII | Ephrin receptors | EPHA1 to EPHA8 EPHB1 to EPHB6 | EFNA1 to EFNA5 EFNB1 to EFNB3 |
Class IX | Récepteurs TAM | AXL MERTK TYRO3 | GAS6 PROS1 |
Class X | LTK/ALK receptors | LTK ALK | PTN MDK |
Class XI | Angiopoietin receptors | TIE1 TEK (TIE2) | – ANGPT1-4 |
Class XII | ROR receptors | ROR1 ROR2 | WNT proteins |
Class XIII | DDR receptors | DDR1 DDR2 | Collagen |
Class XIV | RET receptor | RET | GDNF |
Classes XV–XX | ROS receptor RYK receptor MUSK receptor Lemur receptors PTK7 receptor STYK1 receptor | ROS1 RYK MUSK AATK, LMTK2, LMTK3 PTK7 (no TK activity) STYK1 | ? WNT proteins ? ? WNT proteins ? |
Fig. 1.1
The tyrosine kinase receptors family. Fourteen of the main TKR families are represented schematically. For each receptor, the abbreviated names are indicated below and the ligands above. The various TRKs differ essentially by their extracellular segment which contain characteristic protein domains able to carry out original functions and involved in ligand binding: leucine-rich domains, cysteine-rich domains, immunoglobulin-like domains, cadherin domains, fibronectin III-like domains, EGF-like domains, etc. The intracellular segment especially contains the catalytic tyrosine kinase domain, the C-terminal tyrosine residues serving as phosphorylation substrates and, in the case of ephrin receptors, a PDZ domain
These phosphotyrosine residues are recognised by proteins equipped with binding domains called ‘SH2 domains’ (SRC homology domains 2) and ‘PTB domains’ (phosphotyrosine-binding domains). These proteins are able to recognise in a very specific manner the different phosphorylated tyrosine residues of the activated receptor. More than 100 proteins bearing a SH2 domain and 35 bearing a PTB domain have been identified. Two types of signalling can be implemented:
Cytoplasmic proteins with a SH2 or a PTB domain can be phosphorylated on tyrosine residues, thanks to the catalytic activity of the TKR, and hence display a new biological activity resulting from this phosphorylation. Some of them are cytoplasmic tyrosine kinases, such as SRC (from sarcoma) or JAK (Janus kinase, Chap. 4), tyrosine phosphatases such as SHP1 (SH2 domain-containing phosphatase 1) or other protein types regulated by phosphorylation, such as phospholipase C gamma (PLCγ).
Adapter (docking) proteins, also bearing a SH2 or a PTB domain, can be recruited after TKR activation without subsequent phosphorylation but simply because they can specifically recognise and bind the phosphotyrosine residues of the activated receptor. This is, for instance, the case of GRB2 (growth factor receptor-binding protein 2), which is one of the main entries into the MAP kinase pathway (Chap. 2), and of p85 (gene PIK3R1), which is one of the main entries into the phosphoinositide 3-kinase pathway (Chap. 3).
1.1.2 Oncogenic Alterations
Mutations and overexpressions of growth factors certainly play an important role in oncogenesis, but the primary role of GF molecular alterations in oncogenesis has not often been demonstrated; in contrast, germline inactivating mutations of growth factor genes are responsible for many hereditary diseases such as various forms of dwarfism. In contrast, at the level of TKRs, numerous alterations have been found in cancers which play a major role in oncogenesis. The main alterations will be detailed along the presentation of the different TKR families.
Growth factors and their receptors constitute one of the major research fields to identify pharmacological targets in oncology; the development of targeted therapies has been, for a large part, based upon the research of molecules able to inhibit the proliferation signalling pathways from their first steps. The main successes obtained up to now precisely concern growth factors and tyrosine kinase receptors.
1.1.3 Pharmacological Targets
Growth factors can be targeted with molecules that are able to trap them and block their activity in the extracellular space. Soluble receptors, aptamers, antibodies and peptide constructs can be used to this goal. The major success in this respect concerns a humanised monoclonal antibody, bevacizumab, which binds and inactivates a proangiogenic growth factor, VEGFA; the antiangiogenic activity of this antibody is currently used in the treatment of various cancers in the palliative setting, especially colorectal cancers. Other antibodies directed against various growth factors are presently in development.
Tyrosine kinase receptors can be targeted either at the extracellular level, thanks to monoclonal antibodies, or at the intracellular level by small molecules able to inhibit their tyrosine kinase activity. Several receptors have been thus successfully targeted, especially the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factors (VEGFRs), against which several drugs have been marketed; several other TKRs have been taken in consideration for targeting and appropriate drugs are under development. Whereas monoclonal antibodies are by definition highly specific of a given TKR, this is not so for tyrosine kinase inhibitors (TKIs), which may present a preferential binding on a given receptor but are also active on other TKRs with significant activity. Some TKIs are, therefore, multi-targeting compounds, and it may be sometimes difficult to identify the receptor whose inhibition is actually responsible for pharmacological effect. Table 1.2 presents the main TKIs that have been developed and the target receptor(s) that they inhibit.
Table 1.2
Some tyrosine kinase inhibitors (TKIs)
TKI name | Code number | Pharmaceutical laboratory | Target | Present status |
|---|---|---|---|---|
Gefitinib | ZD1839 | AstraZeneca | EGFR (reversible) | Phase III (Iressa®) |
Erlotinib | OS-774 | Roche | EGFR (reversible) | Approved (Tarceva®) |
Lapatinib | GW572016 | GSK | EGFR, ERBB2 | Approved (Tyverb®) |
Canertinib | CI-1033 | Pfizer | Pan-ERBB (irreversible) | Phase II |
Afatinib | BIBW-2992 | Boehringer Ingelheim | EGFR, ERBB2 (reversible) | Phase III |
Dacomitinib | PF-00299804 | Pfizer | Pan-ERBB (irreversible) | Phase III |
HM781-36B | Hanmi | Pan-ERBB (irreversible) | Phase I | |
Neratinib | HKI-272 | Wyeth | EGFR, ERBB2, (irreversible) | Phase II |
Pelitinib | EKB-569 | Wyeth | EGFR, ERBB2, (irreversible) | Phase II |
Vandetanib | ZD6474 | AstraZeneca | EGFR, FGFR1, VEGFR2, RET | Approved (Zactima® or Caprelsa®) |
Sunitinib | SU-11248 | Pfizer | VEGFR, PDGFR, KIT, FLT3, FGFR1 | Approved (Sutent®) |
Sorafenib | BAY-439006 | Bayer | B-RAF, RET, VEGFR, FGFR1, FLT3 | Approved (Nexavar®) |
Vatalanib | PTK787/ZK222584 | Novartis | VEGFR, KIT | Phase III |
Axitinib | AG-013736 | Pfizer | VEGFR, PDGFR, KIT | ATU (Inlyta®) |
Semaxanib | SU-5416 | Pharmacia | KIT, FLT3, VEGFR2 | Stopped |
Cediranib | AZD-2171 | AstraZeneca | VEGFR, PDGFR | Phase III |
Motesanib | AMG-706 | Amgen | VEGFR, PDGFR, RET, KIT, FLT3 | Phase II |
Pazopanib | GW-786034 | GSK | VEGFR, PDGFR | Phase III |
Imatinib | STI-571 | Novartis | BCR-ABL, KIT, PDGFR | Approved (Glivec®) |
Nilotinib | AMN-107 | Novartis | BCR-ABL, KIT, PDGFR, RET | Approved (Tasigna®) |
Dasatinib | BMS-354825 | BMS | BCR-ABL, KIT, PDGFR | AMM (Sprycel®) |
Masitinib | AB Science | FGFR3, PDGFR, KIT | Phase III | |
Lestaurtinib | CEP-701 | Cephalon | FLT3, NTRK, JAK2 | Phase III |
Crizotinib | PF-02341066 | Pfizer | ALK, MET | Approved (Xalkori®) |
Regorafenib | BAY-73-4506 | Bayer | VEGFR, KIT, RET | Approved (Stivarga®) |
1.2 The Paradigmatic Example of the EGF (ERBB) Family
1.2.1 Growth Factors and Their Receptors
Eleven growth factors of the EGF family have been identified: EGF itself, TGFα (transforming growth factor α), amphiregulin (AREG), epiregulin (EREG), HBEGF (heparin-binding EGF), β-cellulin (BTC), epigen or epithelial mitogen (EPGN) and four neuregulins (NRG1-4). These factors play a major role in the proliferation capacity of many tissues. EGF itself is a small soluble molecule of 53 amino acids, with three intramolecular disulphide bridges (Fig. 1.2a). It derives from a voluminous transmembrane protein of 1,207 amino acids containing a series of analogous egf motifs whose release by cleavage in the extracellular space provides soluble factors. The membrane insertion of native EGF permits its action on adjoining cells, explaining thus a juxtacrine activity. The other factors of the EGF family are also synthesised as transmembrane precursors but contain only one egf extracellular motif. The cleavage of the transmembrane precursors into soluble diffusible growth factors is carried out by metalloproteinases such as TACE (TNF-alpha-converting enzyme) or ADAM17 (a disintegrin and metalloproteinase) (Fig. 1.2b).
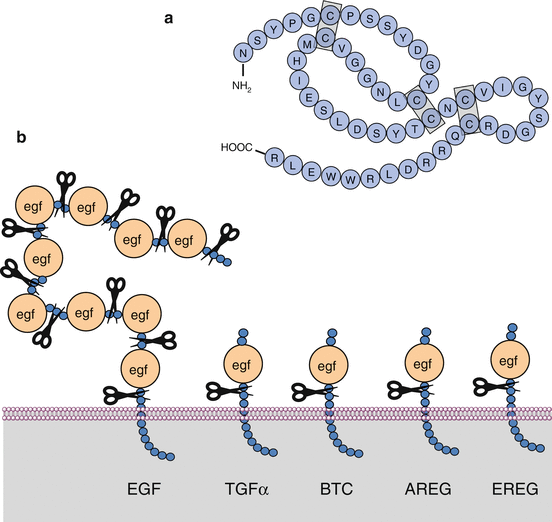
Fig. 1.2
EGF family growth factors. (a) Primary structure of epidermal growth factor (EGF), comprised of 53 amino acids with three intramolecular disulphide bridges. (b) General structure of the ligands of the EGF family. EGF itself is synthesised as a transmembrane precursor bearing 9 egf motifs which can be cleaved by a metalloproteinase such as ADAM17. The other growth factors are also synthesised as transmembrane precursors but contain only one cleavable EGF motif
These 11 growth factors are recognised by 4 distinct ERBB (class I) receptors (EGFR or ERBB1, ERBB2 or HER2, ERBB3, ERBB4). There is a complex combinatorial pattern of interaction between the 11 growth factors and the 4 receptors able to recognise them and activate a signalling pathway. This interaction leads to the homo- or heterodimerisation of two molecules of receptor (Fig. 1.3). EGF, TGFα, amphiregulin and epigen only recognise and bind EGFR itself; HBEGF, epiregulin and β-cellulin recognise and bind EGFR and ERBB4; neuregulins 1 and 2 recognise and bind ERBB3 and ERBB4, and neuregulins 3 and 4 ERBB4. There are two peculiarities: the ERBB2 receptor has no ligand (‘deaf’ receptor) and cannot undergo homodimerisation in normal conditions; the ERBB3 receptor has no tyrosine kinase activity (‘dumb’ receptor) and its homodimers are inactive and cannot transduce a signal.
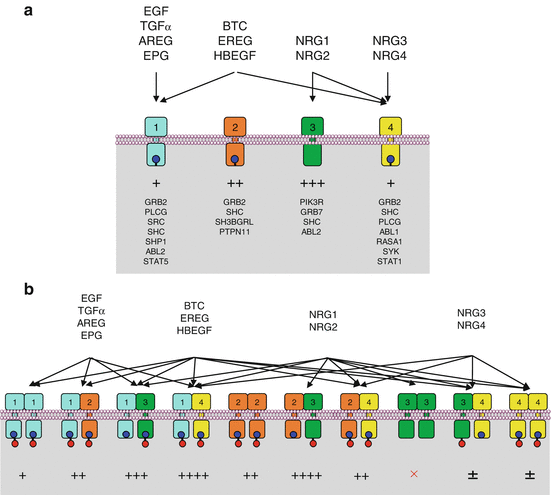
Fig. 1.3
Growth factors and receptors of the ERBB family. The 11 ligands and 4 receptors of the ERBB family are associated in a precise combinatory pattern, according to the respective affinity of each ligand for each receptor. The combinatorial pattern of ligand–receptor interactions is represented in (a) before dimerisation and in (b) after dimerisation (ten possible combinations). The resulting signal is of variable intensity according to the respective affinity of the diverse phosphotyrosine residues of the receptor for the SH2 or PTB domain-containing proteins. The ERBB2 receptor does not recognise any ligand, and its homodimerisation only occurs when it is abundant enough in the membrane, especially due to gene amplification. The ERBB3 receptor displays no kinase activity, and its homodimerisation induces no intracellular signal. The main proteins that recognise the phosphotyrosine residues via their SH2 or PTB domain are indicated below each receptor, and the quantitative importance of the interaction is represented by + signs
1.2.2 ERBB Receptors Activation
EGFR dimerisation is permitted by the unmasking of a binding site between two molecules of receptor, this unmasking being induced by the binding of each molecule of ligand to a molecule of receptor. At the inactive state, the receptor adopts a ‘tethered’ conformation, not allowing the interaction between two molecules of receptor (Fig. 1.4). The binding of EGF, at the level of the extracellular domains 1 and 3, allows the conversion to an ‘extended’ conformation, which unveils a binding site for another molecule of receptor having identically bound a molecule of ligand. The ERBB2 receptor has no ligand because it is always under extended conformation and, therefore, can be dimerised with other receptors without prior activation.
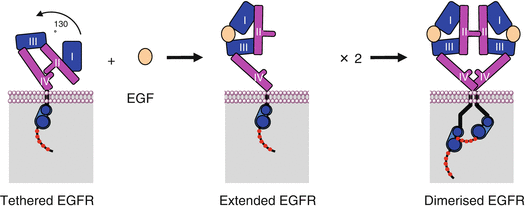
Fig. 1.4
EGFR dimerisation. In the absence of ligand, EGFR adopts a tethered conformation and cannot be dimerised; in the presence of a ligand such as EGF, the conformation of the receptor becomes extended, which unveils an affinity site at the level of the extracellular domain II. Dimerisation induces a change in the relative arrangement of the intracellular catalytic domains of the receptor, allowing autophosphorylation to occur
The dimerisation step allows the contact between two intracellular catalytic domains. Their interaction induces the opening of the active centre of the enzyme and allows ATP to access this centre, thus called the ATP pocket; the catalytic tyrosine kinase activity is then operative. The orientation of the two intracellular domains is asymmetric, one of the catalytic domains being able to phosphorylate the tyrosine residues of the C-terminal end of the second one (Fig. 1.5). The phosphotyrosine residues may then be recognised and bound by a great variety of proteins bearing an SH2 (SRC homology-2) or a PTB (phosphotyrosine-binding) domain. The two types of signalling described above can be then implemented, firstly through the activation of kinases or phosphatases such as SRC or SHP1, which are phosphorylated and activated by the tyrosine kinase activity of the receptor, and secondly through adapter proteins such as GRB2 or p85α (gene PIK3R1). The receptor phosphotyrosine residues that are recognised by each SH2- or PTB-domain-containing protein have been identified: for instance, SRC recognises and binds with high affinity the EGFR phosphotyrosine residue at position Tyr974, SHP1 the residue Tyr1173, GRB2 the residues Tyr1148 and Tyr1173, etc. In addition, the various ligands that activate EGFR induce the phosphorylation of different tyrosine residues: as a result, TGFα or AREG may induce specific effects, distinct from those induced by EGF.
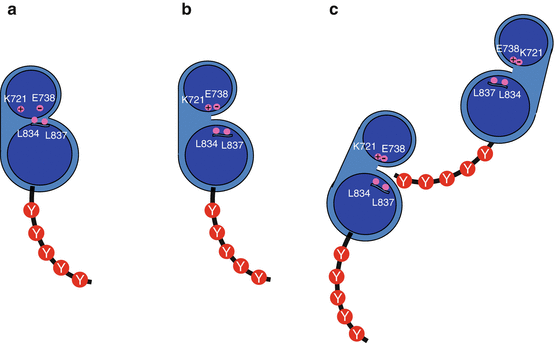
Fig. 1.5
EGFR autophosphorylation. (a) Before receptor activation, the catalytic site is not accessible to ATP because of the presence of the aliphatic amino acid residues Leu833 and Leu838. (b) Upon receptor activation, the catalytic site is open to ATP and ATP can interact with charged amino acid residues (Lys716 and Glu734). (c) The phosphorylation of the C-terminal tyrosine residues (Y) of one of the EGFR monomers is carried out by the catalytic activity of the second one
The various ERBB receptors are activated by their ligands and activate in turn the downstream effectors according to the same general mechanisms, involving homo- or heterodimerisation and autophosphorylation. In addition to the various combinations already mentioned (homodimers and heterodimers) and the various growth factors that induce their activation, Fig. 1.3 presents the main proteins that are activated by each of the receptors and the intensity of the resulting signal. Since proteins with SH2 or PTB domains have variable affinities for the different phosphotyrosine residues of the receptors, the nature and the intensity of the signal vary according to the dimer that has been formed, the tyrosine residues that have been phosphorylated and the SH2- or PTB-domain-containing proteins available in the cell.
ERBB receptors are internalised by endocytosis and are subsequently either recycled or deactivated by transfer into the lysosome. Ligand binding accelerates internalisation and decreases thus the availability of the receptor at the surface of the cell. Internalisation occurs within clathrin-containing vesicles and can be followed, after transfer into endosomes, by ubiquitinylation by an E3 ubiquitin ligase (see Annex C) called CBL (Casitas B-lineage lymphoma), which displays a binding domain for phosphotyrosine residues.
1.2.3 Oncogenic Alterations
The amplification of the EGFR and ERBB2 genes are oncogenic events; for EGFR, this amplification occurs in several types of epithelial cancers, but its importance in oncogenesis is still discussed; for ERBB2, an amplification occurs in 15–25 % of breast cancers and constitutes a characteristic oncogenic event, defining the ERBB2-positive cancers and allowing specific targeted therapies. ERBB2 amplification also occurs in gastric cancers and some other malignancies. The mechanism of gene amplification remains incompletely understood.
Two types of oncogenic mutations can be found in the EGFR gene. The first one concerns the extracellular part of the protein, a part of which is deleted, leading to a permanent activation of the receptor in the absence of ligand. The vIII mutation is the most frequent one (with a large deletion of amino acids 24–297) and is found especially in glioblastoma. The second type concerns the intracellular catalytic site of the receptor (tyrosine kinase activity). These mutations have been identified essentially in a small proportion (5–10 %) of non-small-cell lung cancers and allow the permanent accessibility of the catalytic site to ATP: they permit the autophosphorylation of the tyrosine residues in the absence of a mitogenic signal and of receptor dimerisation. These mutations are found on exon 18 (G719X, 5 % of activating mutations), exon 19 (various deletions in codons 747–751, 45 %), exon 20 (rare insertions) and exon 21 (L858R, 50 %, the most frequent one). Since they maintain the ATP-binding domain open, they are also responsible for the sensitivity of tumour cells to inhibitors of the tyrosine kinase activity of the receptor, being thus simultaneously oncogenic and drug-sensitising mutations.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


