G6PD is a housekeeping gene expressed in all cells. Glucose-6-phosphate dehydrogenase (G6PD) is part of the pentose phosphate pathway, and its main physiologic role is to provide NADPH. G6PD deficiency, one of the commonest inherited enzyme abnormalities in humans, arises through one of many possible mutations, most of which reduce the stability of the enzyme and its level as red cells age. G6PD-deficient persons are mostly asymptomatic, but they can develop severe jaundice during the neonatal period and acute hemolytic anemia when they ingest fava beans or when they are exposed to certain infections or drugs. G6PD deficiency is a global health issue.
Key points
- •
Glucose-6-phosphate dehydrogenase (G6PD) deficiency, expressed in red cells, is mostly asymptomatic; however, G6PD-deficient persons develop acute hemolytic anemia (AHA) when exposed to fava beans, to infection, or to certain drugs, including primaquine.
- •
The gene encoding G6PD maps to the X chromosome. Therefore, full-blown G6PD deficiency is more common in males, but female heterozygotes are also at risk of hemolysis.
- •
G6PD deficiency is widespread in the entire world and its epidemiology correlates with that of malaria; different mutant alleles underlie G6PD deficiency in different populations.
- •
Primaquine is still the only drug that can eradicate Plasmodium vivax hypnozoites; to promptly prevent or to treat hemolytic anemia, it is important to test for G6PD before administering primaquine.
Introduction
G6PD was discovered and biochemically characterized in 1932 by Otto Warburg and Walter Christian in yeast and in red cells as an enzyme with a redox function. It was one of the first enzymes of glucose metabolism to be identified, but, although Warburg did not know that, the clinical manifestations of what later became known as G6PD deficiency had been already described. In the nineteenth century, pediatricians in Greece, Portugal, and Italy observed severe anemia and hemoglobinuria in children who had eaten fava beans – hence, the term favism ; it was noticed that favism tended to recur in the same persons and also that it ran in families. Subsequently, since the 1920s, it was observed that an adverse side effect of 8-aminoquinolines (primaquine and plasmoquine), used for the treatment and the prophylaxis of malaria, was AHA. No connection to favism was suspected at the time, but again it was reported that it was only in certain people that this side effect occurred, and in those people it could happen again on rechallenge with the same drug; this became known as the primaquine sensitivity syndrome .
In 1956, Paul Carson’s group in Chicago reported that red cells from primaquine-sensitive persons were deficient in G6PD (enzyme activity <15% of normal), and in 1958 Gennaro Sansone’s group in Genoa, Italy, found the same deficiency in children with a previous history of favism. It was promptly proved that G6PD deficiency was genetically determined and that its inheritance was X-linked. Almost as soon as Mary Lyon discovered the X-chromosome inactivation phenomenon in mice, Ernie Beutler’s group found independently, using G6PD as a marker, that the same applied to humans. At the time this was the first example of a hemolytic anemia due to an inherited abnormality expressed in red cells; hence, the term enzymopathy was coined, in analogy to hemoglobinopathy. Reassuringly, however, it was clear that in the absence of an exogenous trigger, G6PD-deficient persons had no pathology; hence, primaquine-induced or fava bean–induced AHA became a prototype of a disease arising from a specific interaction between a gene and an environmental factor, just at the time when the term pharmacogenetics was coined.
At approximately the same time, Tony Allison and Arno Motulsky hypothesized that genetically determined G6PD deficiency might have been favored by malaria selection; this spurred a flurry of studies aiming to determine the frequency of this trait in many countries. It quickly emerged that G6PD deficiency was widespread in human populations in all continents; a wealth of epidemiologic data were tabulated by David Livingstone as early as 1967. In the meantime, the World Health Organization (WHO) Human Genetics, then headed by Italo Barrai, was prompt in taking on board the public health implications of such a widespread genetic abnormality; in 1966, a study group was arranged with the remit to review available data and to agree on a measure of standardization for the study of G6PD deficiency.
This article focuses on the essentials of G6PD deficiency as a global health problem and on the essentials of its clinical manifestations, which are a paradigmatic example of a highly specific interaction between an inherited abnormality and exogenous agents that trigger hemolysis. Space does not permit a comprehensive coverage, particularly with respect to management, for which existing literature is referred to.
Introduction
G6PD was discovered and biochemically characterized in 1932 by Otto Warburg and Walter Christian in yeast and in red cells as an enzyme with a redox function. It was one of the first enzymes of glucose metabolism to be identified, but, although Warburg did not know that, the clinical manifestations of what later became known as G6PD deficiency had been already described. In the nineteenth century, pediatricians in Greece, Portugal, and Italy observed severe anemia and hemoglobinuria in children who had eaten fava beans – hence, the term favism ; it was noticed that favism tended to recur in the same persons and also that it ran in families. Subsequently, since the 1920s, it was observed that an adverse side effect of 8-aminoquinolines (primaquine and plasmoquine), used for the treatment and the prophylaxis of malaria, was AHA. No connection to favism was suspected at the time, but again it was reported that it was only in certain people that this side effect occurred, and in those people it could happen again on rechallenge with the same drug; this became known as the primaquine sensitivity syndrome .
In 1956, Paul Carson’s group in Chicago reported that red cells from primaquine-sensitive persons were deficient in G6PD (enzyme activity <15% of normal), and in 1958 Gennaro Sansone’s group in Genoa, Italy, found the same deficiency in children with a previous history of favism. It was promptly proved that G6PD deficiency was genetically determined and that its inheritance was X-linked. Almost as soon as Mary Lyon discovered the X-chromosome inactivation phenomenon in mice, Ernie Beutler’s group found independently, using G6PD as a marker, that the same applied to humans. At the time this was the first example of a hemolytic anemia due to an inherited abnormality expressed in red cells; hence, the term enzymopathy was coined, in analogy to hemoglobinopathy. Reassuringly, however, it was clear that in the absence of an exogenous trigger, G6PD-deficient persons had no pathology; hence, primaquine-induced or fava bean–induced AHA became a prototype of a disease arising from a specific interaction between a gene and an environmental factor, just at the time when the term pharmacogenetics was coined.
At approximately the same time, Tony Allison and Arno Motulsky hypothesized that genetically determined G6PD deficiency might have been favored by malaria selection; this spurred a flurry of studies aiming to determine the frequency of this trait in many countries. It quickly emerged that G6PD deficiency was widespread in human populations in all continents; a wealth of epidemiologic data were tabulated by David Livingstone as early as 1967. In the meantime, the World Health Organization (WHO) Human Genetics, then headed by Italo Barrai, was prompt in taking on board the public health implications of such a widespread genetic abnormality; in 1966, a study group was arranged with the remit to review available data and to agree on a measure of standardization for the study of G6PD deficiency.
This article focuses on the essentials of G6PD deficiency as a global health problem and on the essentials of its clinical manifestations, which are a paradigmatic example of a highly specific interaction between an inherited abnormality and exogenous agents that trigger hemolysis. Space does not permit a comprehensive coverage, particularly with respect to management, for which existing literature is referred to.
Biochemistry of glucose-6-phosphate dehydrogenase and glucose-6-phosphate dehydrogenase deficiency
G6PD is a housekeeping enzyme, expressed in all cells of the body, that catalyzes the oxidation of glucose 6-phosphate (G6P) to 6-phosphoglucono-δ-lactone ( Fig. 1 ), which is then hydrolyzed to 6-phosphoglucono-∂-lactone this, in turn, through the action of the enzyme phosphogluconate dehydrogenase (6PGD), is further oxidized and decarboxylated to the pentose sugar ribulose 5-phosphate. Both G6PD and 6PGD have NADP as coenzyme, and therefore 2 molecules of NADPH are formed per molecule of G6P oxidized by G6PD (see Fig. 1 ). Because the product of these reactions is pentose, G6PD is commonly referred to as the first enzyme of the pentose phosphate pathway. On the other hand, from targeted inactivation of G6PD in embryonic stem cells and from other lines of evidence it became clear that the prime physiologic role of G6PD is the production of NADPH.
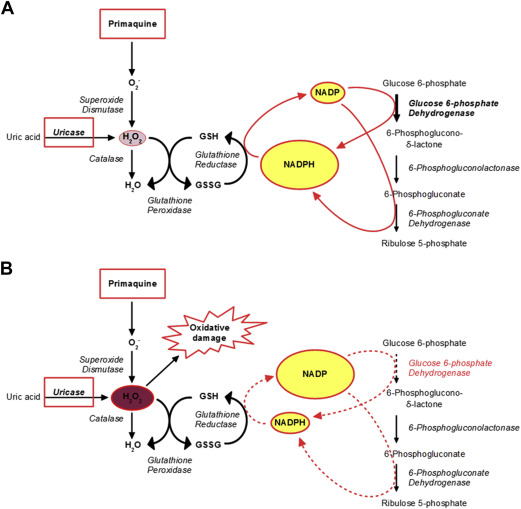
In most cells of the human body NADPH is the key electron donor required for many biosynthetic processes, including several reactions in the pathways of fatty acid synthesis, cholesterol, and steroid hormone synthesis, as well as in the formation from ribose of deoxyribose required for DNA synthesis. In most cells there are several enzymes catalyzing dehydrogenase reactions – other than G6PD – that produce NADPH, and therefore even when G6PD is deficient there may be no shortage of NADPH. The situation is radically different in red blood cells, because the other NADPH-producing enzymes have been sacrificed in the course of erythroid cell differentiation; at the same time, these cells do not need NADPH for the biosynthetic pathways (discussed previously) because they do not exist, having been sacrificed as well. On the other hand, red cells have a great need for the other major function of NADPH: defense against oxidative stress or oxidative attack. This defense is largely mediated through the glutathione cycle, whereby a steady regeneration of reduced glutathione (GSH) depends on a steady supply of NADPH (see Fig. 1 ). Because the red cell is a professional loader, carrier, and unloader of hemoglobin-bound oxygen, and because free radicals can be formed in the process, it is crucially important that it can defend itself against endogenous oxidative stress, even when there is no exogenous attack in sight.
G6PD deficiency is due to inherited mutations in the G6PD gene (discussed later) that are expressed in all cells; and it is already clear from the above that erythrocytes, more than other cells, are vulnerable to the consequences of this defect. But there is an additional important reason for this. As red cells age in circulation, there tends to be a gradual decrease in many of their functions, because individual proteins underlying those functions decay exponentially in these ribosome-less cells that cannot make new protein ; as a result, G6PD activity is approximately 50 times less in a normal red cell that is ready to be removed on day 120 compared with when it itself was a reticulocyte. This process is further magnified with those mutations—the large majority—that compromise the in vivo stability of the G6PD protein.
G6PD deficiency is never complete; if it were complete, it would be lethal. Therefore, in most cases, in the steady state, the consequences of G6PD deficiency are not noticeable (see Fig. 1 ); the NADPH produced by the residual G6PD activity and by 6PGD activity is just enough to keep the red cell going, with marginal reduction of its life span. If an exogenous oxidative stress is applied, however, G6PD-deficient red cells are unable to step up NADPH production (which normal red cells do); as a consequence, GSH is rapidly depleted (see Fig. 1 ), hemoglobin and other proteins are damaged, and eventually the red cell becomes prey to macrophages or hemolyzes altogether.
Molecular-genetic basis of glucose-6-phosphate dehydrogenase deficiency
The G6PD gene consists of 13 exons (the first of which is noncoding) and it encodes a 515–amino acids (AAs) protein subunit, the homodimer of which is enzymatically active; the dimer can further dimerize to give an enzymatically active homotetramer. Each subunit has 1 molecule of tightly bound NADP in addition to binding sites for the NADP substrate and the G6P substrate.
As discussed previously, G6PD deficiency was known from formal genetics to be inherited as an X-linked trait, and the G6PD gene maps to the long arm of the X chromosome (band Xq28). X-linkage has important implications with respect to G6PD deficiency. First, in males there are only 2 genotypes: hemizygous normal and hemizygous G6PD deficient. In females there are 3 genotypes: homozygous normal, homozygous deficient, and heterozygous. Second, although it is often stated that G6PD deficiency is more common in males, this is not correct; according to the fundamental principle of population genetics (the Hardy-Weinberg equilibrium), homozygous females are much more rare than hemizygous males, but heterozygous females are much more numerous ( Table 1 ). A bonus of X-linkage is that male frequencies indicate directly allele frequencies (see Table 1 ). Third, as a result of X-chromosome inactivation, heterozygous females are genetic mosaics ; on average, one-half of their red cells are G6PD normal and one-half are G6PD deficient. There is a wide distribution around this average, whereby in some females the enzyme activity phenotype overlaps with normal, whereas in others it overlaps with the G6PD deficiency as seen in homozygotes ; this has obvious clinical implications.
| Glucose-6-Phosphate Dehydrogenase Deficiency Allele Frequency, q | Glucose-6-Phosphate Dehydrogenase–Deficient Hemizygous Males, % | Glucose-6-Phosphate Dehydrogenase–Deficient Homozygous Females, % | Glucose-6-Phosphate Dehydrogenase–Deficient Heterozygous Females, % |
|---|---|---|---|
| 0.01 | 1 | 0.01 | 2 |
| 0.05 | 5 | 0.25 | 9.5 |
| 0.25 | 25 | 6.25 | 37.5 |
All G6PD mutations known ( Table 2 ), except G6PD A, are associated with more or less severe enzyme deficiency but never with complete loss of activity; there are no frameshift mutations in the database (such mutations presumably are lethal; discussed previously and later), and the only nonsense mutation has been found in a heterozygous woman. The mutations that underlie G6PD deficiency are spread throughout the coding region ( Fig. 2 ). The most recent compilation lists 186 G6PD alleles in addition to the normal or wild-type gene, referred to traditionally as G6PD B.
| Class | Single Nucleotide Substitutions | Multiple Nucleotide Substitutions | Deletions | Intronic Mutations | Total Number | |
|---|---|---|---|---|---|---|
| Polymorphic | II, III, and IV | 27 | 4 | 0 | 0 | 31 |
| Perhaps polymorphic b | II and III | 55 | 6 | 0 | 0 | 61 |
| Nonpolymorphic | I | 69 | 5 | 10 | 1 | 85 |
| Undefined | — | 8 | 0 | 0 | 1 | 9 |
| Total number | — | 159 | 15 | 10 | 2 | 186 |
a G6PD B is regarded as the human wild type and, therefore, is not included in the count.
b Not every G6PD variant in class II or III has been proved polymorphic, but this is probable.
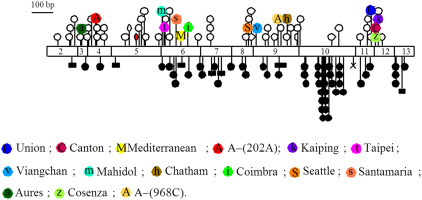
A large majority of mutations (159) are missense mutations due to single nucleotide substitutions causing single AA replacements; however, there are also multiple missense mutations within the same allele as well as in frame deletions and rare mutations that affect splicing. The frequency of single and multiple nucleotide substitutions is approximately the same among variants that are or may be polymorphic and those that are not. In contrast, deletions are only found within the nonpolymorphic variants (see Table 2 ).
Not surprisingly, different mutations cause both quantitative and qualitative changes in the enzyme. This has led to a classification of G6PD variants based on the degree of deficiency and on clinical manifestations ( Table 3 ). A cluster of mutations, most of them in exon 10, encode AAs that are in the dimerization domain ; they affect markedly the stability of the dimer and they produce class I variants (see Table 3 ), causing chronic nonspherocytic hemolytic anemia (CNSHA).
| Current Classification | Proposed Revision | ||||
|---|---|---|---|---|---|
| Class | Residual Glucose-6-Phosphate Dehydrogenase Activity (% of Normal) a | Clinical Manifestations | Examples of Genetic Variants | Class | Residual Glucose-6-Phosphate Dehydrogenase Activity (% of Normal) a |
| I b | <10 c | CNSHA d (NNJ, acute exacerbations) | Guadalajara, Nara, Sunderland | I | <10 |
| II | <10 c | None in the steady state | Mediterranean, Canton, Union | II + III | <30 e |
| III | 10–60 | None in the steady state | A−, Mahidol, Seattle | ||
| IV | 100 | None | A, B | IV | >85 |
| V | >100 | None | — | — | — |
a Levels of residual G6PD activity in hemizygous males.
b The definition of class I variants is not biochemical but clinical (ie, class I variants cause CNSHD).
c The range of G6PD activity is similar in class I and class II variants, which may seem strange because the clinical phenotype is significantly different. It must be considered, however, that (1) in CNSHD, there is always reticulocytosis, which increases G6PD levels, and (2) in some class I variants, the residual G6PD activity may be similar to a class II or even a class III variant, but the enzyme kinetics may be unfavorable.
d When hemolysis is not compensated, chronic anemia is present and blood transfusions may be necessary at times or even at regular intervals.
e Cutoff is indicated as 30%, because all G6PD variants in class II and III described so far have a residual activity of less than 30%.
A remarkable feature at the genomic level is that the G6PD gene overlaps with the IKBKG/NEMO gene, which is transcribed in the opposite direction and mutations of which are responsible for the serious disease incontinentia pigmenti . Some of these mutations are lethal in males, but in females they include large deletions of G6PD , which are compatible with life thanks to selection for cells in which the active X chromosome has an intact G6PD allele.
Evolution
From full genome databases, it is inferred that G6PD is not present in Archaebacteria , which, because they live in environments with low or no oxygen, hardly need defense against oxidative stress. In all other living organisms, G6PD is highly conserved; the AA sequence similarity from microorganisms to mammals ranges from 43% to 98%. This high degree of conservation must mean that the G6PD protein has been shaped by evolution early and robustly to perform its enzymatic function well. In many plants, there are 2 G6PD genes—1 encodes cytosolic G6PD and the other G6PD present in plastids —and a human pseudogene is known.
In the alignment of G6PD coding sequences from all organisms, those of Plasmodia stand out because they have a long 5′ extension, which encodes the metabolically related enzyme 6-phosphoglucono-δ-lactonase. It has been suggested that the protein product of the parasite’s bifunctional gene might be a new target for antimalarials.
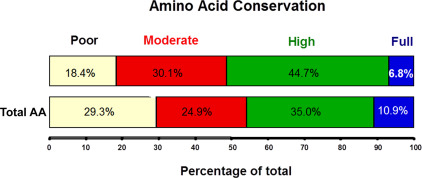
It is often thought that in an enzyme protein, functionally critical AA residues are less likely to change over evolutionary times; as a result, mutations that cause disease in general are more likely to affect the most evolutionarily conserved residues. This simple correlation, however, is not always seen, and it seems that it does not exactly hold for G6PD. An analysis of 103 G6PD mutants causing G6PD deficiency in humans, as against 52 G6PD sequences from 45 different organisms, has shown that most mutations (74%) are in highly and moderately conserved (50%–99% of similarity) AAs, whereas few mutations are in fully conserved or in poorly conserved AAs ( Fig. 3 ). This distinct relationship suggests that mutations in poorly conserved AAs may remain inconspicuous because they do not cause significant G6PD deficiency, whereas mutations in fully conserved AAs might be lethal, which is not surprising in the case of a gene, such as G6PD, present in a single functioning copy and indispensable for life.
Epidemiology and malaria selection
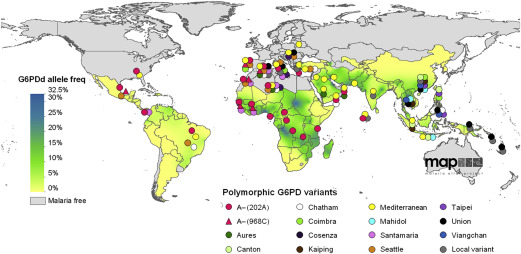
The geographic distribution of G6PD deficiency is spectacular, because it spares no continent ( Fig. 4 ), yet population frequencies are highly variable, because they reflect 2 major factors in the epidemiology of a genetic abnormality: environmental selection and migration. The correlation with the epidemiology of malaria, discussed previously, is obvious, for instance, in areas as distant as tropical Africa, Southeast Asia, and the Vanuatu archipelago in the Pacific; and parts of Southern Europe can be included, where malaria was endemic until 2 to 3 generations ago. G6PD deficiency is also common in the Americas, however, including areas that have never had malaria; this is largely accounted for by migrations, voluntary or otherwise, from Africa, Asia, and Europe. G6PD deficiency has no significant frequency in native American populations.
Related posts:
 The Global Burden of Anemia
The Global Burden of Anemia
 Problems and Approaches for Blood Transfusion in the Developing Countries
Problems and Approaches for Blood Transfusion in the Developing Countries
 Hematological Practice in India
Hematologic Changes Associated with Specific Infections in the Tropics
Global Approach to Hematologic Malignancies
Improving Laboratory and Clinical Hematology Services in Resource Limited Settings
Hematological Practice in India
Hematologic Changes Associated with Specific Infections in the Tropics
Global Approach to Hematologic Malignancies
Improving Laboratory and Clinical Hematology Services in Resource Limited Settings
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


