Abbreviations
- ACTH Adrenocorticotropic hormone
- ADH Antidiuretic hormone (vasopressin)
- APS Autoimmune polyglandular syndrome
- AVP Arginine vasopressin
- CBG Corticosteroid-binding globulin
- CRH Corticotropin-releasing hormone
- DAX1 Dosage-sensitive sex reversal-adrenal hypoplasia gene product
- DHEA Dehydroepiandrosterone
- DOC Deoxycorticosterone
- GH Growth hormone
- GIP Glucose-dependent insulinotropic peptide/Gastrointestinal inhibitory polypeptide
- GnRH Gonadotropin-releasing hormone
- HLA Human leukocyte antigen
- HPA Hypothalamic-pituitary-adrenal
- HPLC High-performance liquid chromatography
- IL Interleukin
- IPSS Inferior petrosal sinus sampling
- IRMA Immunoradiometric assay
- LH Luteinizing hormone
- LPH Lipotropin
- PMN Polymorphonuclear neutrophi
- PRA Plasma renin activity
- PRL Prolactin
- PTH Parathyroid hormone
- SF-1 Steroidogenic factor-1a
- SHBG Sex hormone–binding globulin
- StAR Steroidogenic acute regulatory protein
- TRH Thyrotropin-releasing hormone
- TSH Thyroid-stimulating hormone (thyrotropin)
- VLCFA Very long chain fatty acid
Glucocorticoids and Adrenal Androgens: Introduction
The adrenal cortex produces many steroid hormones of which the most important are cortisol, aldosterone, and the adrenal androgens. Disorders of the adrenal glands lead to classic endocrinopathies such as Cushing syndrome, Addison disease, hyperaldosteronism, and the syndromes of congenital adrenal hyperplasia. This chapter describes the physiology and disorders of the glucocorticoids and the adrenal androgens. Disorders of aldosterone secretion are discussed in Chapter 10 and congenital defects in adrenal hormone biosynthesis in Chapters 10 and 14. Hirsutism and virilization (which reflect excess androgen action) are discussed in Chapter 13.
Advances in diagnostic procedures have simplified the evaluation of adrenocortical disorders; in particular, the assay of plasma glucocorticoids, androgens, and adrenocorticotropin (ACTH) has allowed more rapid and precise diagnosis. In addition, advances in surgical and medical treatment have improved the outlook for patients with these disorders.
Embryology and Anatomy
The adrenal cortex is of mesodermal origin and derives from a single cell lineage characterized by expression of certain transcription factors such as steroidogenic factor-1 (SF-1). At 2 months’ gestation, the cortex, already identifiable as a separate organ, is composed of a fetal zone and a definitive zone similar to the adult adrenal cortex. The adrenal cortex then increases rapidly in size; at mid gestation, it is considerably larger than the kidney and much larger than the adult gland in relation to total body mass. The fetal zone makes up the bulk of the weight of the adrenal cortex at this time. Several genes encoding transcription factors are important in adrenal development and differentiation. These include SF-1 and the product of the dosage-sensitive sex reversal–adrenal hypoplasia gene (DAX1), among others; mutations of the DAX1 gene are associated with congenital adrenal hypoplasia.
The fetal adrenal is under the control of ACTH by mid pregnancy, but the fetal zone is deficient in the activity of 3β-hydroxysteroid dehydrogenase (see section on biosynthesis of cortisol and adrenal androgens, later) and thus produces mainly dehydroepiandrosterone (DHEA) and DHEA sulfate, which serve as precursors of maternal-placental estrogen production after conversion in the liver to 16α-hydroxylated derivatives. The definitive zone synthesizes a number of steroids and is the major site of fetal cortisol synthesis. Mutations of the ACTH- or melanocortin-2 receptor gene are associated with familial glucocorticoid deficiency.
The anatomic relationship of the fetal and definitive zones is maintained until birth, at which time the fetal zone gradually disappears, with a consequent decrease in adrenocortical weight in the 3 months following delivery. During the next 3 years, the adult adrenal cortex develops from cells of the outer layer of the cortex and differentiates into the three adult zones: glomerulosa, fasciculata, and reticularis–adrenal zonation.
The adult adrenal glands, with a combined weight of 8 to 10 g, lie in the retroperitoneum above or medial to the upper poles of the kidneys (Figure 9–1). A fibrous capsule surrounds the gland; the cortex comprises 90% of the adrenal weight, the inner medulla is about 10%.
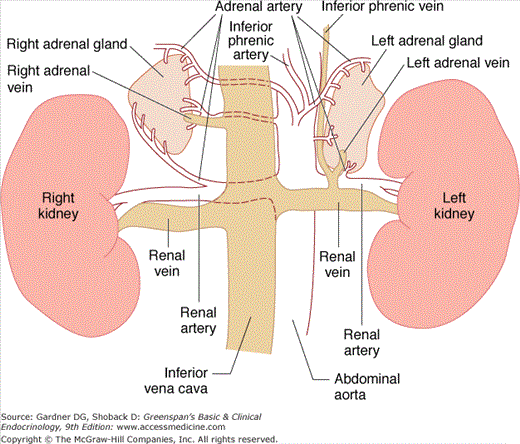
The adrenal cortex is richly vascularized and receives its main arterial supply from branches of the inferior phrenic artery, the renal arteries, and the aorta. These small arteries form an arterial plexus beneath the capsule and then enter a sinusoidal system that penetrates the cortex and medulla, draining into a single central vein in each gland. The right adrenal vein drains directly into the posterior aspect of the vena cava; the left adrenal vein enters the left renal vein. These anatomic features account for the fact that it is relatively easier to catheterize the left adrenal vein than it is to catheterize the right adrenal vein.
Histologically, the adult cortex is composed of three zones: an outer zona glomerulosa, a zona fasciculata, and an inner zona reticularis (Figure 9–2). However, the inner two zones appear to function as a unit (see later). The zona glomerulosa, which produces aldosterone and constitutes about 15% of adult cortical volume, is deficient in 17α-hydroxylase activity and thus cannot produce cortisol or androgens (see later and Chapter 10). The zona glomerulosa lacks a well-defined structure, and the small lipid-poor cells are scattered beneath the adrenal capsule. The zona fasciculata is the thickest layer of the adrenal cortex, making up about 75% of the cortex, and produces cortisol and androgens. The cells of the zona fasciculata are larger and contain more lipid and thus are termed clear cells. These cells extend in columns from the narrow zona reticularis to either the zona glomerulosa or to the capsule. The inner zona reticularis surrounds the medulla and also produces cortisol and androgens. The compact cells of this narrow zone lack significant lipid content but do contain lipofuscin granules. The zonae fasciculata and reticularis are regulated by ACTH; excess or deficiency of this hormone alters their structure and function. Thus, both zones atrophy when ACTH is deficient; when ACTH is present in excess, hyperplasia and hypertrophy of these zones occur. In addition, chronic stimulation with ACTH leads to a gradual depletion of the lipid from the clear cells of the zona fasciculata at the junction of the two zones; these cells thus attain the characteristic appearance of the compact reticularis cells. With chronic excessive stimulation, the compact reticularis cells extend outward and may reach the outer capsule. It is postulated that the zona fasciculata cells can respond acutely to ACTH stimulation with increased cortisol production, whereas the reticularis cells maintain basal glucocorticoid secretion and that induced by prolonged ACTH stimulation.
Figure 9–2
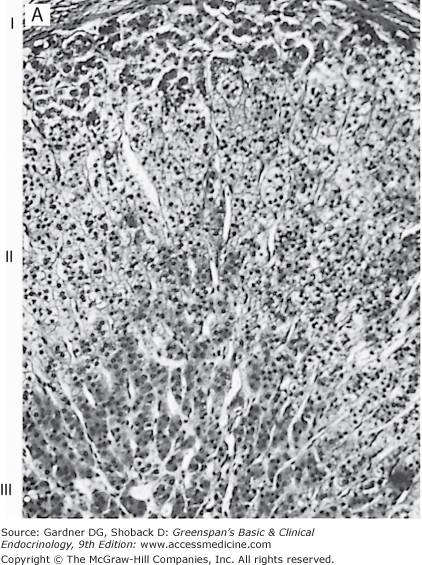
Photomicrograph of the adrenal cortex (H&E stain). A. A low-power general view. I, the glomerulosa; II, the fasciculata; III, the reticularis.
(Reproduced, with permission, from Junqueira LC, Carneiro J. Basic Histology. 7th ed. McGraw-Hill; 1992.)
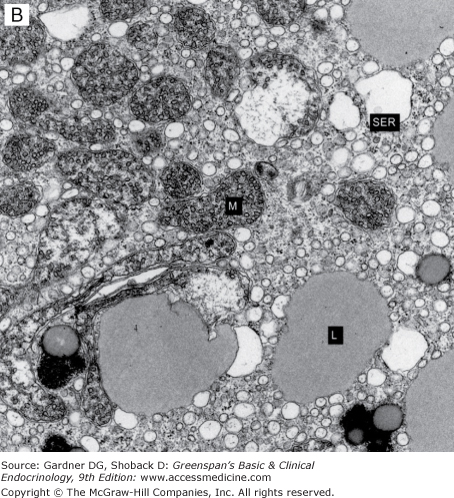
B. Electron micrograph of a normal adrenocortical steroid-producing cell (L, lipid vacuole; M, large mitochondria with tubular cristae; SER, smooth endoplasmic reticulum). (Courtesy of Dr. Medhat O. Hasan, Pathology Department, Louis Stokes Cleveland Department of Veterans Affairs Medical Center.)
Biosynthesis of Cortisol and Adrenal Androgens
The major hormones secreted by the adrenal cortex are cortisol, the androgens, and aldosterone. The carbon atoms in the steroid molecule are numbered as shown in Figure 9–3, and the major biosynthetic pathways and hormonal intermediates are illustrated in Figures 9–4 and 9–5.
Figure 9–3
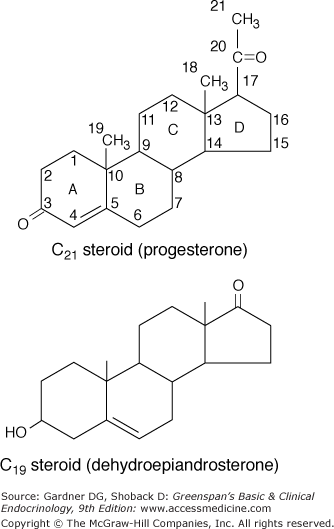
Structure of adrenocortical steroids. The letters in the formula for progesterone identify the A, B, C, and D rings; the numbers show the positions in the basic C21steroid structure. The angular methyl groups (positions 18 and 19) are usually indicated simply by straight lines, as in the lower formula. Dehydroepiandrosterone is a 17-ketosteroid formed by cleavage of the side chain of the C21steroid 17-hydroxypregnenolone and its replacement by an O atom.
(Reproduced, with permission, from Ganong WF. Review of Medical Physiology. 14th ed. McGraw-Hill; 1989.)
Figure 9–4
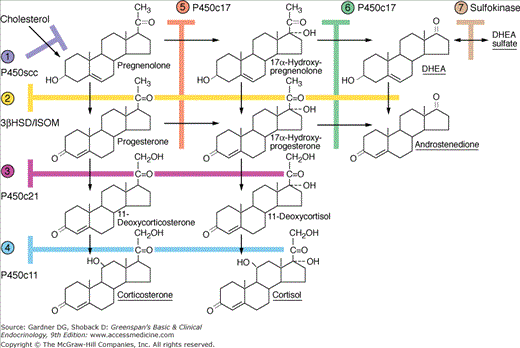
Steroid biosynthesis in the zona fasciculata and zona reticularis of the adrenal cortex. The major secretory products are underlined. The enzymes for the reactions are numbered on the left and at the top of the chart, with the steps catalyzed shown by the shaded bars. ① P450scc, cholesterol 20,22-hydroxylase:20,22 desmolase activity; ② 3βHSD/ISOM, 3-hydroxysteroid dehydrogenase; δ5-oxosteroid isomerase activity; ③ P450c21, 21α-hydroxylase activity; ④ P450c11, 11β-hydroxylase activity; ⑤ P450c17, 17α-hydroxylase activity; ⑥ P450c17, 17,20-lyase/desmolase activity; ⑦ sulfokinase.
(Modified and reproduced, with permission, from Ganong WF. Review of Medical Physiology. 16th ed. McGraw-Hill; 1993.)
Figure 9–5
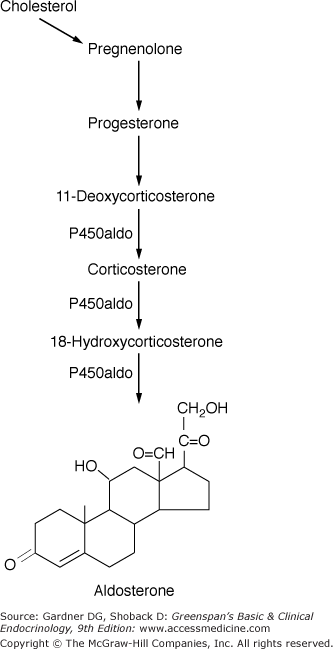
Steroid biosynthesis in the zona glomerulosa. The steps from cholesterol to 11-deoxycorticosterone are the same as in the zona fasciculata and zona reticularis. However, the zona glomerulosa lacks 17α-hydroxylase activity and thus cannot produce cortisol. Only the zona glomerulosa can convert corticosterone to 18-hydroxycorticosterone and aldosterone. The single enzyme P450aldo catalyzes the conversion of 11-deoxycorticosterone to corticosterone to 18-hydroxycorticosterone to aldosterone.
(Modified and reproduced, with permission, from Ganong WF. Review of Medical Physiology. 16th ed. McGraw-Hill; 1993.)
The scheme of adrenal steroidogenic synthesis has been clarified by analysis of the steroidogenic enzymes. Most of these enzymes belong to the family of cytochrome P450 oxygenases (see Table 9–1 for current and historical nomenclature conventions). In mitochondria, the CYP11A gene, located on chromosome 15, encodes P450scc, the enzyme responsible for cholesterol side chain cleavage. CYP11B1, a gene located on chromosome 8, encodes P450c11, another mitochondrial enzyme, which mediates 11β-hydroxylation in the zona reticularis and zona fasciculata. This reaction converts 11-deoxycortisol to cortisol and 11-deoxycorticosterone (11-DOC) to corticosterone. In the zona glomerulosa, CYP11B2, also located on chromosome 8, encodes the enzyme P450aldo, also known as aldosterone synthase. P450aldo mediates 11β-hydroxylation, 18-hydroxylation, and 18-oxidation to convert 11-DOC to corticosterone, 18-hydroxycorticosterone, and aldosterone, respectively. In the endoplasmic reticulum, gene CYP17, located on chromosome 10, encodes a single enzyme, P450c17, which mediates both 17α-hydroxylase activity and 17,20-lyase activity, and the gene CYP21A2 encodes the enzyme P450c21, which mediates 21-hydroxylation of both progesterone and 17-hydroxyprogesterone. The 3β-hydroxysteroid dehydrogenase: δ5,4-isomerase activities are mediated by a single non-P450 microsomal enzyme (see Figure 9–4).
| Enzyme | Gene | Chromosomal Location | Enzyme Activity (or Activities) | Subcellular Localization | Characteristic Features of Normal Tissue-Specific Expression |
|---|---|---|---|---|---|
| Steroidogenic acute regulatory protein (StAR) | StAR | 8p11.2 | Activation of peripheral type benzodiazepine receptor | Outer mitochondrial membrane | All steroid hormone–producing cells except the placenta, Schwann cells, and the brain |
| Peripheral type benzodiazepine receptor | PDA | 22q13.31 | Regulated cholesterol channel | Forms channel at the contact sites between the outer and inner mitochondrial membranes | All steroid hormone–producing cells |
| P450scc | CYP11A | 15q23-24 | Cholesterol-20,22-desmolase | Matrix side of inner mitochondrial membrane | All steroid hormone–producing cells |
| 3β-Hydroxysteroid dehydro genase (HSD)/isomerase | 3β-Hydroxysteroid dehydrogenase type I | 1p13, the HSD3B1 and HSD3B2 loci are located 1-2 cm from the centromeric marker D1Z5 | 3β-Hydroxysteroid dehydrogenase | Smooth endoplasmic reticulum | Expressed in syncytiotrophoblast cells, sebaceous glands |
| 3β-Hydroxysteroid dehydrogenase type II | Expressed in the definitive adrenal cortex and the gonads. Absent from the fetal zone of the adrenal cortex | ||||
| P450c21 | CYP21B | 6p21.3 (close to HLA locus) | 21-hydroxylase | Smooth endoplasmic reticulum | Only in adrenal cortex (all zones); low levels in fetal zone |
| CYP21A | 6p21.3 (close to HLA locus) | Pseudogene | N/A | N/A | |
| P450c11 | CYP11B1 | 8q21-22 | 11β-hydroxylase | Matrix side of inner mitochondrial membrane | Only expressed in zona fasciculata and zona reticularis of the adrenal cortex; low levels in fetal zone |
| P450aldo | CYP11B2 | 8q24.3 | Aldosterone synthase: 11β-hydroxylase, 18-hydroxylase, 18-oxidase | Matrix side of inner mitochondrial membrane | Only expressed in the zona glomerulosa of the adrenal cortex; low levels in fetal zone |
| P450c17 | CYP17 | 10q24-25 | 17α-hydroxylase, 17,20-lyase | Smooth endoplasmic reticulum | Absent from the zona glomerulosa in adults, the placenta, and the definitive zone of fetal adrenal cortex until the third trimester |
| P450arom | CYP19 | 15q | Aromatase | Smooth endoplasmic reticulum | Expressed in numerous tissues including gonads, brain, adrenal, adipose tissue, and bone |
Because of enzymatic differences between the zona glomerulosa and the inner two zones, the adrenal cortex functions as two separate units, with differing regulation and secretory products. Thus, the zona glomerulosa, which produces aldosterone, lacks 17α-hydroxylase activity and cannot synthesize 17α-hydroxypregnenolone and 17α-hydroxyprogesterone, which are the precursors of cortisol and the adrenal androgens. The synthesis of aldosterone by this zone is primarily regulated by the renin-angiotensin system and by potassium (see Chapter 10).
The zona fasciculata and zona reticularis (see Figure 9–4) produce cortisol, androgens, and small amounts of estrogens. These zones, primarily regulated by ACTH, do not express the gene CYP11B2 (encoding P450aldo) and therefore cannot convert 11-DOC to aldosterone (see Chapter 10).
Synthesis of cortisol and the androgens by the zonae fasciculata and reticularis begins with cholesterol, as does the synthesis of all steroid hormones. Plasma lipoproteins are the major source of adrenal cholesterol, although synthesis within the gland from acetate also occurs. Low-density lipoprotein accounts for about 80% of cholesterol delivered to the adrenal gland. A small pool of free cholesterol within the adrenal is available for rapid synthesis of steroids when the adrenal is stimulated. When stimulation occurs, there is also increased hydrolysis of stored cholesteryl esters to free cholesterol, increased uptake from plasma lipoproteins, and increased cholesterol synthesis within the gland. The acute response to a steroidogenic stimulus is mediated by the steroidogenic acute regulatory protein (StAR). This mitochondrial phosphoprotein enhances cholesterol transport from the outer to the inner mitochondrial membrane. Mutations in the StAR gene result in congenital lipoid adrenal hyperplasia with severe cortisol and aldosterone deficiencies at birth.
The conversion of cholesterol to pregnenolone is the rate-limiting step in adrenal steroidogenesis and the major site of ACTH action in the adrenal. This step occurs in the mitochondria and involves two hydroxylations and then the side chain cleavage of cholesterol. A single enzyme, CYP11A, mediates this process; each step requires molecular oxygen and a pair of electrons. The latter are donated by NADPH to adrenodoxin reductase (ferredoxin reductase), a flavoprotein, and then to adrenodoxin, an iron-sulfur protein, and finally to CYP11A. Both adrenodoxin reductase and adrenodoxin are also involved in the action of CYP11B1 (see earlier). Electron transport to microsomal cytochrome P450 involves P450 reductase, a flavoprotein distinct from adrenodoxin reductase. Pregnenolone is then transported outside the mitochondria before further steroid synthesis occurs.
Cortisol synthesis proceeds by 17α-hydroxylation of pregnenolone by CYP17 within the smooth endoplasmic reticulum to form 17α-hydroxypregnenolone. This steroid is then converted to 17α-hydroxyprogesterone after conversion of its 5,6 double bond to a 4,5 double bond by the 3β-hydroxysteroid dehydrogenase: δ5,4-oxosteroid isomerase enzyme complex, which is also located within the smooth endoplasmic reticulum. An alternative but apparently less important pathway in the zonae fasciculata and reticularis is from pregnenolone to progesterone to 17α-hydroxyprogesterone (see Figure 9–4).
The next step, which is again microsomal, involves the 21-hydroxylation by CYP21A2 of 17α-hydroxyprogesterone to form 11-deoxycortisol; this compound is further hydroxylated within mitochondria by 11β-hydroxylation (CYP11B1) to form cortisol. The zona fasciculata and zona reticularis also produce 11-DOC, 18-hydroxydeoxycorticosterone, and corticosterone. However, as noted above, the absence of the mitochondrial enzyme CYP11B2 prevents production of aldosterone by these zones of the adrenal cortex (Figure 9–5). Cortisol secretion under basal (ie, nonstressed) conditions ranges from 8 to 25 mg/d (22-69 μ mol/d), with a mean of about 9.2 mg/d (25 μ mol/d)—rates lower than most previous calculations.
The production of adrenal androgens from pregnenolone and progesterone requires prior 17α-hydroxylation (CYP17) and thus does not occur in the zona glomerulosa. The major quantitative production of androgens is by conversion of 17α-hydroxypregnenolone to the 19-carbon compounds (C-19 steroids) DHEA and its sulfate conjugate DHEA sulfate. Thus, 17α-hydroxypregnenolone undergoes removal of its two-carbon side chain at the C17position by microsomal 17,20-desmolase (CYP17), yielding DHEA with a keto group at C17. DHEA is then converted to DHEA sulfate by a reversible adrenal sulfokinase. The other major adrenal androgen, androstenedione, is produced mostly from DHEA, mediated by CYP17, and possibly from 17α-hydroxyprogesterone, also by CYP17. Androstenedione can be converted to testosterone, although adrenal secretion of this hormone is minimal. The adrenal androgens, DHEA, DHEA sulfate, and androstenedione, have minimal intrinsic androgenic activity, and they contribute to androgenicity by their peripheral conversion to the more potent androgens testosterone and dihydrotestosterone. Although DHEA and DHEA sulfate are secreted in greater quantities, androstenedione is qualitatively more important, because it is more readily converted peripherally to testosterone (see Chapter 12). Of note, recent studies have identified de novo synthesis of some steroid hormones in nerve and cardiac tissues, where they appear to act as paracrine or autocrine factors. Steroidogenic enzymes (eg, 3β-hydroxysteroid dehydrogenase and aromatase) are expressed in many tissues.
ACTH is the trophic hormone of the zonae fasciculata and reticularis and the major regulator of cortisol and adrenal androgen production, although other factors produced within the adrenal, including neurotransmitters, neuropeptides, and nitric oxide also play a role. ACTH in turn is regulated by the hypothalamus and central nervous system via neurotransmitters and corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP). The neuroendocrine control of CRH and ACTH secretion involves three mechanisms (see later and Chapter 4).
ACTH administration leads to the rapid synthesis and secretion of steroids; plasma levels of these hormones rise within minutes. ACTH increases RNA, DNA, and protein synthesis. Chronic stimulation leads to adrenocortical hyperplasia and hypertrophy; conversely, ACTH deficiency results in decreased steroidogenesis and is accompanied by adrenocortical atrophy, decreased gland weight, and decreased protein and nucleic acid content.
ACTH binds to high-affinity plasma membrane receptors, thereby activating adenylyl cyclase and increasing cyclic adenosine monophosphate, which in turn activates intracellular phosphoprotein kinases (Figure 9–6), including StAR. ACTH action results in increased free cholesterol formation as a consequence of increased cholesterol esterase activity and decreased cholesteryl ester synthetase as well as increased lipoprotein uptake by the adrenal cortex. This process stimulates the rate-limiting step—cholesterol delivery to the side chain cleavage enzyme (P450scc or CYP11A1) for conversion to δ5-pregnenolone, thereby initiating steroidogenesis.
Figure 9–6
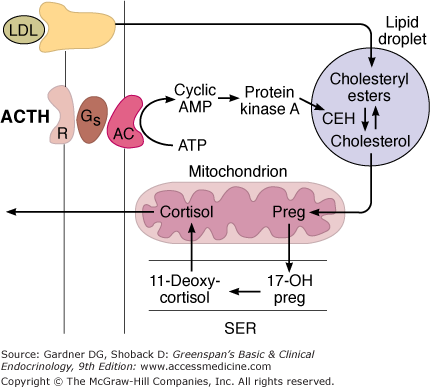
Mechanism of action of ACTH on cortisol-secreting cells in the inner two zones of the adrenal cortex. When ACTH binds to its receptor (R), adenylyl cyclase (AC) is activated via Gs. The resulting increase in cAMP activates protein kinase A, and the kinase phosphorylates cholesteryl ester hydrolase (CEH), increasing its activity. Consequently, more free cholesterol is formed and converted to pregnenolone in the mitochondria. Note that in the subsequent steps in steroid biosynthesis, products are shuttled between the mitochondria and the smooth endoplasmic reticulum (SER).
(Reproduced, with permission, from Ganong WF. Review of Medical Physiology. 16th ed. McGraw-Hill; 1993.)
Cortisol secretion is closely regulated by ACTH, and plasma cortisol levels parallel those of ACTH (Figure 9–7). There are three mechanisms of neuroendocrine control: (1) episodic secretion and the circadian rhythm of ACTH, (2) stress responsiveness of the hypothalamic-pituitary-adrenal (HPA) axis, and (3) feedback inhibition by cortisol of ACTH secretion.
Figure 9–7
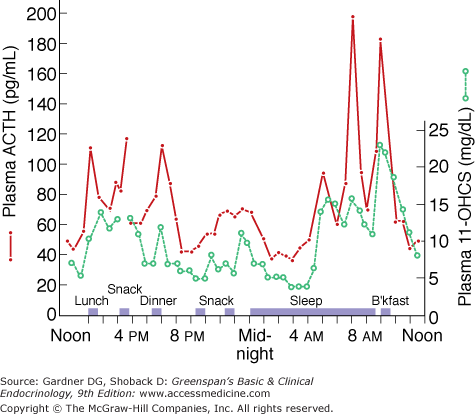
Fluctuations in plasma ACTH and glucocorticoids (11-OHCS) throughout the day. Note the greater ACTH and glucocorticoid rises in the morning before awakening.
(Reproduced, with permission, from Krieger DT, et al. Characterization of the normal temporal pattern of corticosteroid levels. J Clin Endocrinol Metab. 1971;32:266.)
Circadian rhythm—Circadian rhythm is superimposed on episodic secretion; it is the result of central nervous system events that regulate both the number and magnitude of CRH and ACTH secretory episodes. Cortisol secretion is low in the late evening and continues to decline in the first several hours of sleep, at which time plasma cortisol levels may be undetectable. During the third and fifth hours of sleep there is an increase in secretion; but the major secretory episodes begin in the sixth to eighth hours of sleep (see Figure 9–7) and then begin to decline as wakefulness occurs. About half of the total daily cortisol output is secreted during this period. Cortisol secretion then gradually declines during the day, with fewer secretory episodes of decreased magnitude; however, there is increased cortisol secretion in response to eating and exercise.
Although this general pattern is consistent, there is considerable intra- and interindividual variability, and the circadian rhythm may be altered by changes in sleep pattern, light-dark exposure, and feeding times. The rhythm is also changed by (1) physical stresses such as major illness, surgery, trauma, or starvation; (2) psychologic stress, including severe anxiety, endogenous depression, and the manic phase of manic-depressive psychosis; (3) central nervous system and pituitary disorders; (4) Cushing syndrome; (5) liver disease and other conditions that affect cortisol metabolism; (6) chronic renal failure; and (7) alcoholism. Cyproheptadine inhibits the circadian rhythm, possibly by its antiserotonergic effects, whereas other drugs usually have no effect.
Stress responsiveness—Plasma ACTH and cortisol secretion are also characteristically responsive to physical stress. Thus, plasma ACTH and cortisol are secreted within minutes following the onset of stresses such as surgery and hypoglycemia, and these responses abolish circadian periodicity if the stress is prolonged. Stress responses originate in the central nervous system and increase hypothalamic CRH and thus pituitary ACTH secretion. Stress responsiveness of plasma ACTH and cortisol is abolished by prior high-dose glucocorticoid administration and in spontaneous Cushing syndrome; conversely, the responsiveness of ACTH secretion is enhanced following adrenalectomy. Regulation of the HPA axis is linked to that of the immune system. For example, interleukin-1 (IL-1) stimulates ACTH secretion, and cortisol inhibits IL-1 synthesis.
Feedback inhibition—The third major regulator of ACTH and cortisol secretion is that of feedback inhibition by glucocorticoids of CRH, ACTH, and cortisol secretion. Glucocorticoid feedback inhibition occurs at both the pituitary and hypothalamus and involves two distinct mechanisms—fast and delayed feedback inhibition.
Fast feedback inhibition of ACTH secretion is rate-dependent; that is, it depends on the rate of increase of the glucocorticoid but not the dose administered. This phase is rapid (within minutes) and transient (lasting <10 minutes), suggesting mediation by a noncytosolic glucocorticoid receptor mechanism. Delayed feedback inhibition is both time- and dose-dependent. With continued glucocorticoid administration, ACTH levels continue to decrease and become unresponsive to stimulation, ultimately resulting in suppression of CRH and ACTH release and atrophy of the zonae fasciculata and reticularis. The suppressed HPA axis fails to respond to stress and stimulation. Delayed feedback appears to act via the classic glucocorticoid receptor mechanism (see later).
Adrenal androgen production in adults is also regulated by ACTH; both DHEA and androstenedione exhibit circadian periodicity in concert with ACTH and cortisol. In addition, plasma concentrations of DHEA and androstenedione increase rapidly with ACTH administration and are suppressed by glucocorticoid administration. DHEA sulfate, because of its slow metabolic clearance rate, does not exhibit a diurnal rhythm. The existence of a separate anterior pituitary hormone that regulates adrenal androgen secretion has long been postulated but not yet proved.
Circulation of Cortisol and Adrenal Androgens
Cortisol and the adrenal androgens circulate bound to plasma proteins. The plasma half-life of cortisol (60-90 minutes) is determined by the extent of plasma binding and by the rate of metabolic inactivation.
Cortisol and adrenal androgens are secreted in an unbound state; however, these hormones bind to plasma proteins on entering the circulation. Cortisol binds mainly to corticosteroid-binding globulin (CBG, transcortin) and to a lesser extent to albumin, whereas the androgens bind chiefly to albumin. Bound steroids are biologically inactive; the unbound or free fraction is active. The plasma proteins may provide a pool of circulating cortisol by delaying metabolic clearance, thus preventing more marked fluctuations of plasma-free cortisol levels during episodic secretion by the gland. Because there are no binding proteins in saliva, salivary cortisol reflects free cortisol.
Under basal conditions, about 10% of the circulating cortisol is free, about 75% is bound to CBG, and the remainder is bound to albumin. The plasma free cortisol level is approximately 1 μ g/dL, and it is this biologically active cortisol that is regulated by ACTH.
Corticosteroid-binding globulin (CBG) has a molecular weight of about 50,000, is produced by the liver, and binds cortisol with high affinity. The CBG in plasma has a cortisol-binding capacity of about 25 μ g/dL. When total plasma cortisol concentrations rise above this level, the free concentration rapidly increases and exceeds its usual fraction of 10% of the total cortisol. Other endogenous steroids usually do not appreciably affect cortisol binding to CBG; an exception is in late pregnancy, when progesterone may occupy about 25% of the binding sites on CBG. Synthetic steroids do not bind significantly to CBG—with the exception of prednisolone. CBG levels are increased in high-estrogen states (pregnancy; estrogen or oral contraceptive use), hyperthyroidism, diabetes, certain hematologic disorders, and on a genetic basis. CBG concentrations are decreased in familial CBG deficiency, hypothyroidism, and protein deficiency states such as severe liver disease or nephrotic syndrome.
Albumin has a much greater capacity for cortisol binding but a lower affinity. It normally binds about 15% of the circulating cortisol, and this proportion increases when the total cortisol concentration exceeds the CBG-binding capacity. Synthetic glucocorticoids are extensively bound to albumin (eg, about 75% of dexamethasone in plasma is bound to albumin).
Androstenedione, DHEA, and DHEA sulfate circulate weakly bound to albumin. However, testosterone is bound extensively to a specific globulin, sex hormone–binding globulin (SHBG) (see Chapter 12).
Metabolism of Cortisol and Adrenal Androgens
The metabolism of these steroids renders them inactive and increases their water solubility, as does their subsequent conjugation with glucuronide or sulfate groups. These inactive conjugated metabolites are more readily excreted by the kidney. The liver is the major site of steroid catabolism and conjugation, and 90% of these metabolized steroids are excreted by the kidney.
Cortisol is modified extensively before excretion in urine; less than 1% of secreted cortisol appears in the urine unchanged.
Hepatic metabolism of cortisol involves a number of metabolic conversions of which the most important (quantitatively) is the irreversible inactivation of the steroid by δ4-reductases, which reduce the 4,5 double bond of the A ring. Dihydrocortisol, the product of this reaction, is then converted to tetrahydrocortisol by a 3-hydroxysteroid dehydrogenase. Cortisol is also converted extensively by 11β-hydroxysteroid dehydrogenase to the biologically inactive cortisone, which is then metabolized by the enzymes described above to yield tetrahydrocortisone. Tetrahydrocortisol and tetrahydrocortisone can be further altered to form the cortoic acids. These conversions result in the excretion of approximately equal amounts of cortisol and cortisone metabolites. Cortisol and cortisone are also metabolized to the cortols and cortolones and to a lesser extent by other pathways (eg, to 6β-hydroxycortisol).
More than 95% of cortisol and cortisone metabolites are conjugated by the liver and then reenter the circulation to be excreted in the urine. Conjugation is mainly with glucuronic acid at the 3α-hydroxyl position.
The metabolism of cortisol is altered by a number of circumstances. It is decreased in infants and in the elderly. It is impaired in chronic liver disease, leading to decreased renal excretion of cortisol metabolites; however, the plasma cortisol level remains normal. Hypothyroidism decreases both metabolism and excretion; conversely, hyperthyroidism accelerates these processes. Cortisol clearance may be reduced in starvation and anorexia nervosa and is also decreased in pregnancy because of the elevated CBG levels. The metabolism of cortisol to 6β-hydroxycortisol is increased in the neonate, in pregnancy, with estrogen therapy, and in patients with liver disease or severe chronic illness. Cortisol metabolism by this pathway is also increased by drugs that induce hepatic microsomal enzymes, including barbiturates, phenytoin, mitotane, aminoglutethimide, and rifampin. These alterations generally are of minor physiologic importance as the free cortisol levels remain relatively stable in these conditions. However, they result in decreased excretion of the urinary metabolites of cortisol measured as 17-hydroxycorticosteroids. These conditions and drugs have a greater influence on the metabolism of synthetic glucocorticoids and may result in inadequate plasma levels of the administered glucocorticoid because of rapid clearance and metabolism.
Aldosterone is the principal mineralocorticoid controlling sodium and potassium exchange in the distal nephron. Mineralocorticoid receptors in the kidney are responsible for this effect, and the sensitivities of both the glucocorticoid receptor and the mineralocorticoid receptor for cortisol in vitro are similar. Small changes in aldosterone affect sodium and potassium exchange in the kidney, whereas free and biologically active cortisol does not, yet cortisol circulates in much higher concentrations. This apparent paradox is explained by an intracellular enzyme—11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2)—that metabolizes cortisol to the inactive cortisone and protects the mineralocorticoid receptor from cortisol binding (Figure 9–8). However, when circulating cortisol is extremely high (as in severe Cushing syndrome), this prereceptor metabolism of cortisol is overwhelmed and the mineralocorticoid receptor is activated by cortisol, resulting in volume expansion, hypertension, and hypokalemia. The active ingredient of licorice (glycyrrhizic acid) actually inhibits 11β-HSD2 and gives cortisol free access to the unprotected mineralocorticoid receptor in the kidney, causing hypokalemia and hypertension. In addition, some tissues can actually convert the inactive cortisone to cortisol with the isoform called 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1). The skin expresses this enzyme, explaining why cortisone cream can be effective. More importantly, the liver expresses 11β-HSD1 and can activate cortisone to cortisol, thereby completing the cortisol-cortisone shunt such that the kidney inactivates cortisol to cortisone and the liver can reactivate cortisone to cortisol. The expression of 11β-HSD1 in adipose tissue may contribute to abdominal obesity seen in metabolic syndrome without biochemical hypercortisolism.
Figure 9–8

Cortisol-cortisone shunt. Contrasting functions of the isozymes of 11β-HSD. 11β-HSD2 is an exclusive 11β-dehydrogenase that acts in classical aldosterone target tissues to exclude cortisol from otherwise nonselective mineralocorticoid receptors. Inactivation of cortisol also occurs in placenta. 11β-HSD1 is a predominant 11β-reductase in vivo that acts in many tissues to increase local intracellular glucocorticoid concentrations and thereby maintain adequate exposure of relatively low affinity glucocorticoid receptors to their ligand.
Adrenal androgen metabolism results either in degradation and inactivation or the peripheral conversion of these weak androgens to their more potent derivatives testosterone and dihydrotestosterone. DHEA is readily converted within the adrenal to DHEA sulfate, the adrenal androgen secreted in greatest amount. DHEA secreted by the gland is also converted to DHEA sulfate by the liver and kidney, or it may be converted to δ4-androstenedione. DHEA sulfate may be excreted without further metabolism; however, both it and DHEA are also metabolized to 7α- and 16α-hydroxylated derivatives and by 17β reduction to δ5-androstenediol and its sulfate. Androstenedione is converted either to testosterone or by reduction of its 4,5 double bond to etiocholanolone or androsterone, which may be further converted by 17α reduction to etiocholanediol and androstanediol, respectively. Testosterone is converted to dihydrotestosterone in androgen-sensitive tissues by 5β reduction, and it, in turn, is mainly metabolized by 3α reduction to androstanediol. The metabolites of these androgens are conjugated either as glucuronides or sulfates and excreted in the urine.
Biologic Effects of Adrenal Steroids
Although glucocorticoids were originally so called because of their influence on glucose metabolism, they are currently defined as steroids that exert their effects by binding to specific cytosolic receptors that mediate the actions of these hormones. These glucocorticoid receptors are present in virtually all tissues, and glucocorticoid-receptor interaction is responsible for most of the known effects of these steroids. Alterations in the structure of the glucocorticoids have led to the development of synthetic compounds with greater glucocorticoid activity. The increased activity of these compounds is due to increased affinity for the glucocorticoid receptors and delayed plasma clearance, which increases tissue exposure. In addition, many of these synthetic glucocorticoids have negligible mineralocorticoid effects and thus do not result in sodium retention, hypertension, and hypokalemia. This section describes the molecular mechanisms of glucocorticoid action and the effects on individual metabolic functions and tissues (Table 9–2).
| Target System | Specific Target | Physiologic Function | Cushing Disease | Addison Disease |
|---|---|---|---|---|
| Intermediary metabolism | Liver | Increased expression of gluconeogenic enzymes, phosphoenolpyruvate carboxykinase, glucose-6-phosphatase, and fructose-2,6-bisphosphatase | Increased hepatic glucose output; together with insulin, increased hepatic glycogen stores | Diminished hepatic glucose output and glycogen stores |
| Adipose tissue | Permissive for lipolytic signals (catecholamines, GH) leading to elevated plasma FFA to fuel gluconeogenesis | Overall effect (together with insulin): central obesity (truncal obesity, moon facies, and buffalo hump) | Decreased adiposity and decreased lipolysis | |
| Skeletal muscle | Degradation of fibrillar muscle proteins by activating the ubiquitin pathway, thereby providing amino acid substrates for gluconeogenesis | Muscle weakness and wasting mainly in proximal muscles; increased urinary nitrogen excretion (urea from amino acids) | Muscle weakness, decreased muscle glycogen stores; decreased urinary nitrogen excretion | |
| Plasma glucose | Maintains plasma glucose during fasting (antihypoglycemic action); increases plasma glucose during stress (hyperglycemic action) | Impaired glucose tolerance, insulin-resistant diabetes mellitus; increased plasma glucose is due to decreased peripheral glucose utilization and increased hepatic glucose output | Hypoglycemia, increased insulin sensitivity | |
| Calcium homeostasis | Kidney | Decreased reabsorption of calcium | Hypercalciuria without hypercalcemia leading to secondary hyperparathyroidism | Retardation of bone growth mainly through decreased GH; hypercalcemia possible |
| Bone, cartilage | Inhibition of collagen synthesis and bone deposition | Retardation of bone growth and bone age by direct action and by decreasing GH; osteoporosis in adults | ||
| Gastrointestinal tract | Inhibition of calcium, magnesium, and phosphate absorption by antagonizing calcitriol actions | |||
| Other endocrine systems | Hypothalamus, pituitary | Decreases endogenous opioid production; depresses gonadotroph responsiveness to GnRH; stimulates GH gene expression by the pituitary; inhibits GH secretion via the hypothalamus | Scanty menses due to suppressed gonadotroph sensitivity to GnRH; suppressed GH secretion by hypothalamic action; minimal suppression of the TRH-TSH axis | Scanty menses by upregulated CRH-endogenous opioid pathway-mediated suppression of GnRH; suppressed GH secretion; hypothyroidism (if present) is due to autoimmune mechanism |
| Pancreas | Inhibits insulin secretion by decreasing the efficacy of cytoplasmic Ca2+ on the exocytotic process | Absolute hyperinsulinemia with relative hypoinsulinemia (lower plasma insulin than expected for the degree of hyperglycemia) | Absolute hypoinsulinemia with relative hyperinsulinemia | |
| Adrenal medulla | Increases PNMT expression and activity (epinephrine synthesis) | Increased responses to sympathoadrenal activation | Decreased responses to sympathoadrenal activation | |
| Carrier proteins (CBG, SHBG, TBG) | Decreases all major hormone-binding proteins | Decrease in total T4, free T4 remains normal | ||
| Immune system | Thymus, lymphocytes | Causes age-related involution of the thymus; induces thymic atrophy | Immunocompromised state; lymphocytopenia | Relative lymphocytosis in peripheral blood |
| Monocytes | Inhibits monocyte proliferation and antigen presentation; decreased production of IL-1, IL-6, and TNFα | Monocytopenia in peripheral blood | Monocytosis in peripheral blood | |
| Granulocytes | Demargination of neutrophils by suppressing the expression of adhesion molecules | Peripheral blood: granulocytosis, eosinopenia | Peripheral blood: granulocytopenia, eosinophilia | |
| Inflammatory response | Inhibition of inflammation by inhibiting PLA2, thereby inhibiting production of leukotrienes and prostaglandins; suppresses COX-2 expression | |||
| Erythrocytes | No significant effect | Increased hemoglobin and hematocrit are due to ACTH-mediated overproduction of androgens | Anemia is more pronounced in women and is due to loss of adrenal androgens; anemia may be related to direct autoimmune targeting of gastric parietal cells | |
| Skin and connective tissue | Antiproliferative for fibroblasts and keratinocytes | Easy bruisability due to dermal atrophy; striae or sites of increased tension, especially sites of adipose tissue accumulation; poor wound healing; hirsutism and acne are due to ACTH-mediated increase of adrenal androgens; hyperpigmentation is a direct effect of ACTH on melanocortin 1 receptors | Darkening of the skin is due to ACTH-mediated stimulation of epidermal melanocortin 1 receptors; vitiligo may occur due to direct autoimmune destruction of melanocytes in circumscribed areas | |
| Breast | Mammary epithelium | Mandatory requirement for lactation | Cushing disease may be associated with galactorrhea | Addison disease is not associated with galactorrhea |
| Lung | Type II alveolar cell | Stimulation of surfactant production | ||
| Cardiovascular system | Heart | Increased contractility | Hypertension | Lower peripheral resistance; hypertension with further postural decrease in blood pressure (orthostatic hypotension); low-voltage ECG |
| Vasculature | Increased vascular reactivity to vasoconstrictors (catecholamines, angiotensin II) | |||
| Na+, K+, and ECF volume | Kidney | Increased GFR and nonphysiologic actions on mineralocorticoid receptors | Hypokalemic alkalosis, increased ECF volume due to mineralocorticoid activity (increased DOC, saturation of type 2 11β-hydroxysteroid dehydrogenase by high levels of cortisol) | Hyponatremia, hyperkalemic acidosis, and decreased ECF volume are mainly due to loss of mineralocorticoid activity |
| Posterior pituitary | Hyponatremia due to SIADH | Increased ADH mainly via hypovolemia-related baroreceptor mechanism | ||
| Psychiatric parameters of CNS function | Mood | Eucortisolemia maintains emotional balance | Initially, euphoria; long-term, depression, psychosis | Depression |
| Appetite | Increases appetite | Hyperphagia | Decreased appetite in spite of improved taste and smell | |
| Sleep | Suppression of REM sleep | Sleep disturbances | ||
| Memory | Sensitizes hippocampal glutamate receptors, induces atrophy of dendrites | Impaired memory, bilateral hippocampal atrophy | ||
| Eye | Increasing intraocular pressure | Cataract formation; increased intraocular pressure | Decreased intraocular pressure |
Glucocorticoid action is initiated by entry of the steroid into the cell and binding to the cytosolic glucocorticoid–receptor proteins (see Figures 1–13, 1–14, and 1–15). The most abundant cytoplasmic glucocorticoid–receptor complex includes two subunits of the 90-kDa heat shock protein (hsp) 90. After binding, the hsp90 subunits dissociate and activated hormone–receptor complexes enter the nucleus and interact with nuclear chromatin acceptor sites. The DNA-binding domain of the receptor is a cysteine-rich region which, when it chelates zinc, assumes a conformation called a zinc finger. The receptor–glucocorticoid complex acts via two mechanisms: (1) binding to specific sites in nuclear DNA, the glucocorticoid regulatory elements; and (2) interactions with other transcription factors such as nuclear factor κB, an important regulator of cytokine genes. These result in altered expression of specific genes and the transcription of specific mRNAs. The resulting proteins elicit the glucocorticoid response, which may be inhibitory or stimulatory depending on the specific gene and tissue affected. Although glucocorticoid receptors are similar in many tissues, the proteins synthesized in response to glucocorticoids vary widely and are the result of expression of specific genes in different cell types. The mechanisms underlying this specific regulation are unknown. Analyses of cloned complementary DNAs for human glucocorticoid receptors have revealed marked structural and amino acid sequence homology between glucocorticoid receptors and receptors for other steroid hormones (eg, mineralocorticoids, estrogen, progesterone) as well as for thyroid hormone and the oncogene v-erb-A. Although the steroid-binding domain of the glucocorticoid receptor confers specificity for glucocorticoid binding, glucocorticoids such as cortisol and corticosterone bind to the mineralocorticoid receptor with an affinity equal to that of aldosterone. Mineralocorticoid receptor specificity is maintained by the expression of 11β-HSD in classic mineralocorticoid-sensitive tissues—the cortisol-cortisone shunt (see earlier).
Although glucocorticoid–receptor complexes and their subsequent regulation of gene expression are responsible for most glucocorticoid effects, other effects may occur through plasma membrane receptors.
The study of glucocorticoid receptors has led to the definition of glucocorticoid agonists and antagonists. These studies have also identified a number of steroids with mixed effects termed partial agonists, partial antagonists, or partial agonist–partial antagonists. In addition, novel glucocorticoid receptor ligands are being developed that have more selectivity in terms of receptor binding or transcription of specific genes.
In humans, cortisol, synthetic glucocorticoids (eg, prednisolone, dexamethasone), corticosterone, and aldosterone are glucocorticoid agonists. The synthetic glucocorticoids have substantially higher affinity for the glucocorticoid receptor, and these have greater glucocorticoid activity than cortisol when present in equimolar concentrations. Corticosterone and aldosterone have substantial affinity for the glucocorticoid receptor; however, their plasma concentrations are normally much lower than that of cortisol, and thus these steroids do not have significant physiologic glucocorticoid effects.
Glucocorticoid antagonists bind to the glucocorticoid receptors but do not elicit the nuclear events required to cause a glucocorticoid response. These steroids compete with agonist steroids such as cortisol for the receptors and thus inhibit agonist responses. Other steroids have partial agonist activity when present alone (ie, they elicit a partial glucocorticoid response). However, in sufficient concentration, they compete with agonist steroids for the receptors and thus competitively inhibit agonist responses; that is, these partial agonists may function as partial antagonists in the presence of more active glucocorticoids. Steroids such as progesterone, 11-deoxycortisol, DOC, testosterone, and 17β-estradiol have antagonist or partial agonist-partial antagonist effects; however, the physiologic role of these hormones in glucocorticoid action is probably negligible, because they circulate in low concentrations. The antiprogestational agent mifepristone has substantial glucocorticoid antagonist properties and has been used to block glucocorticoid action in patients with Cushing syndrome.
Glucocorticoids in general inhibit DNA synthesis. In addition, in most tissues they inhibit RNA and protein synthesis and accelerate protein catabolism. These actions provide substrate for intermediary metabolism; however, accelerated catabolism also accounts for the deleterious effects of glucocorticoids on muscle, bone, connective tissue, and lymphatic tissues. In contrast, RNA and protein synthesis in liver is stimulated.
Glucocorticoids increase hepatic gluconeogenesis by stimulating the gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose-6-phosphatase. They have a permissive effect in that they increase hepatic responsiveness to the gluconeogenic hormone glucagon, and they also increase the release of substrates for gluconeogenesis from peripheral tissues, particularly muscle. This latter effect may be enhanced by the glucocorticoid-induced reduction in peripheral amino acid uptake and protein synthesis. Glucocorticoids also increase glycerol and free fatty acid release by lipolysis and increase muscle lactate release. They enhance hepatic glycogen synthesis and storage by stimulating glycogen synthetase activity and to a lesser extent by inhibiting glycogen breakdown. These effects are insulin dependent.
Glucocorticoids also alter carbohydrate metabolism by inhibiting peripheral glucose uptake in muscle and adipose tissue. This effect and the others described earlier may result in increased insulin secretion in states of chronic glucocorticoid excess.
In adipose tissue, the predominant effect is increased lipolysis with release of glycerol and free fatty acids. This is partially due to direct stimulation of lipolysis by glucocorticoids, but it is also contributed to by decreased glucose uptake and enhancement by glucocorticoids of the effects of lipolytic hormones. Although glucocorticoids are lipolytic, increased fat deposition is a classic manifestation of glucocorticoid excess. This paradox may be explained by the increased appetite caused by high levels of these steroids and by the lipogenic effects of the hyperinsulinemia that occurs in this state. The reason for abnormal fat deposition and distribution in states of cortisol excess is unknown. In these instances, fat is classically deposited centrally in the face, cervical area, trunk, and abdomen; the extremities are usually spared.
The effects of the glucocorticoids on intermediary metabolism can be summarized as follows:
- (1) Effects are minimal in the fed state. However, during fasting, glucocorticoids contribute to the maintenance of plasma glucose levels by increasing gluconeogenesis and decreasing uptake of glucose by adipose tissue.
- (2) Hepatic glucose production is enhanced, as is hepatic RNA and protein synthesis.
- (3) The effects on muscle are catabolic (ie, decreased glucose uptake and metabolism, decreased protein synthesis, and increased release of amino acids). This provides amino acid substrates for gluconeogenesis in the liver.
- (4) In adipose tissue, lipolysis is stimulated. This increases fatty acid delivery to the liver where their metabolism provides energy to support gluconeogenesis.
- (5) In glucocorticoid deficiency, hypoglycemia may result, whereas in states of glucocorticoid excess there may be hyperglycemia, hyperinsulinemia, muscle wasting, and weight gain with abnormal fat distribution.
Glucocorticoids in excess inhibit fibroblasts, lead to loss of collagen and connective tissue, and thus result in thinning of the skin, easy bruising, stria formation, and poor wound healing.
The physiologic role of glucocorticoids in bone metabolism and calcium homeostasis is unknown; however, in excess, they have major deleterious effects. Glucocorticoids directly inhibit bone formation by decreasing cell proliferation and the synthesis of RNA, protein, collagen, and hyaluronate. Initially, supraphysiologic doses of glucocorticoids also stimulate bone resorption, at least in part via interference of the receptor activator of nuclear factor kappa B (RANK)-ligand/RANK/osteoprotegerin pathway. This leads to osteolysis and increased biochemical markers of bone turnover. In addition, glucocorticoids may potentiate the proresorptive actions of parathyroid hormone (PTH) and 1,25-dihydroxycholecalciferol (1,25[OH]2D3) on bone, and this may further contribute to net bone resorption. Chronically, however, the key effect to promote bone loss is due to the detrimental impact on bone formation (see Chapter 8).
Glucocorticoids also have other major effects on mineral homeostasis. They markedly reduce intestinal calcium absorption, which tends to lower serum calcium. This theoretically promotes a state of secondary hyperparathyroidism to maintain the serum calcium within the normal range. In reality, however, chronic elevations in intact PTH levels have been difficult to demonstrate consistently in patients on glucocorticoid therapy. The mechanism of decreased intestinal calcium absorption is thought to result from antagonism of 1,25(OH)2D action in the intestine. It is not due to decreased synthesis or decreased serum levels of the active vitamin D metabolites; in fact, 1,25(OH)2D levels are normal or even increased in the presence of glucocorticoid excess. Increased 1,25(OH)2D synthesis in this setting may result from decreased serum phosphorus levels (see later), increased PTH secretion, direct stimulation by glucocorticoids of renal 1α-hydroxylase, and or reduced target cell (intestine) responsiveness. Glucocorticoids increase urinary calcium excretion, and hypercalciuria is a frequent feature of cortisol excess. They also reduce the tubular reabsorption of phosphate, leading to phosphaturia and decreased serum phosphorus concentrations.
Thus, glucocorticoids in excess result in negative calcium balance, with decreased calcium absorption and increased urinary calcium excretion. Serum calcium levels are maintained, but at the expense of net bone resorption. Decreased bone formation and increased resorption ultimately result in the disabling osteoporosis that is often a major complication of spontaneous and iatrogenic glucocorticoid excess (see Chapter 8).
Glucocorticoids accelerate the development of a number of systems and organs in fetal and differentiating tissues, although the mechanisms are unclear. As discussed above, glucocorticoids are generally inhibitory, and these stimulatory effects may be due to glucocorticoid interactions with other growth factors. Examples of these development-promoting effects are increased surfactant production in the fetal lung and the accelerated development of hepatic and gastrointestinal enzyme systems.
Glucocorticoids in excess inhibit growth in children, and this adverse effect is a major complication of therapy. This may be a direct effect on bone cells, although decreased growth hormone (GH) secretion and insulin-like growth factor I generation also contribute (see Chapter 6).
Erythrocytes—Glucocorticoids have little effect on erythropoiesis and hemoglobin concentration. Although mild polycythemia and anemia may be seen in Cushing syndrome and Addison disease, respectively, these alterations are more likely to be secondary to altered androgen metabolism.
Leukocytes—Glucocorticoids influence both leukocyte movement and function. Thus, glucocorticoid administration increases the number of intravascular polymorphonuclear neutrophils/leukocytes (PMNs) by increasing PMN release from bone marrow, by increasing the circulating half-life of PMNs, and by decreasing PMN movement out of the vascular compartment. Glucocorticoid administration reduces the number of circulating lymphocytes, monocytes, and eosinophils, mainly by increasing their movement out of the circulation. The converse (ie, neutropenia, lymphocytosis, monocytosis, and eosinophilia) is seen in adrenal insufficiency. Glucocorticoids also decrease the migration of inflammatory cells (PMNs, monocytes, and lymphocytes) to sites of injury, and this is probably a major mechanism of the anti-inflammatory actions and increased susceptibility to infection that occur following chronic administration. Glucocorticoids also decrease lymphocyte production and the mediator and effector functions of these cells.
Immunologic effects—Glucocorticoids influence multiple aspects of immunologic and inflammatory responsiveness, including the mobilization and function of leukocytes, as discussed earlier. They inhibit phospholipase A2, a key enzyme in the synthesis of prostaglandins. This inhibition is mediated by a class of peptides called lipocortins or annexins. They also impair the release of effector substances such as the lymphokine IL-1, antigen processing, antibody production and clearance, and other specific bone marrow–derived and thymus-derived lymphocyte functions. The immune system, in turn, affects the HPA axis; IL-1 stimulates the secretion of CRH and ACTH. Although traditionally used as anti-inflammatory and/or immunosuppressive agents, glucocorticoids, especially at lower doses, also have stimulatory and permissive effects on the inflammatory response to injury.
Glucocorticoids may increase cardiac output, and they also increase peripheral vascular tone, possibly by augmenting the effects of other vasoconstrictors (eg, the catecholamines). Glucocorticoids also regulate expression of adrenergic receptors. Thus, refractory shock may occur when the glucocorticoid-deficient individual is subjected to stress. Glucocorticoids in excess may cause hypertension independently of their mineralocorticoid effects. Although the incidence and the precise cause of this problem are unclear, it is likely that the mechanism involves the renin-angiotensin system; glucocorticoids regulate renin substrate, angiotensinogen, the precursor of angiotensin I.
These steroids affect water and electrolyte balance by actions mediated either by mineralocorticoid receptors (sodium retention, hypokalemia, and hypertension) or via glucocorticoid receptors (increased glomerular filtration rate due to increased cardiac output or due to direct renal effects on salt and water retention). Thus, corticosteroids such as betamethasone or dexamethasone, which have little mineralocorticoid activity, increase sodium and water excretion. Glucocorticoid-deficient subjects have decreased glomerular filtration rates and are unable to excrete a water load. This may be further aggravated by increased ADH secretion, which may occur in glucocorticoid deficiency.
Glucocorticoids readily enter the brain, and although their physiologic role in central nervous system function is unknown, their excess or deficiency may profoundly alter behavior and cognitive function.
Excessive glucocorticoids—In excess, the glucocorticoids initially cause euphoria; however, with prolonged exposure, a variety of psychologic abnormalities occur, including irritability, emotional lability, and depression. Hyperkinetic or manic behavior is less common; overt psychoses occur in a small number of patients, particularly those with underlying bipolar disorder. Many patients also note impairment in cognitive functions, most commonly memory and concentration. Other central effects include increased appetite, decreased libido, and insomnia, with decreased rapid eye movement sleep and increased stage II sleep.
Decreased glucocorticoids—Patients with Addison disease are apathetic and depressed and tend to be irritable, negativistic, and reclusive. They have decreased appetite but increased sensitivity of taste and smell mechanisms.
Thyroid function—Glucocorticoids in excess affect thyroid function. Thyroid-stimulating hormone (TSH) synthesis and release are inhibited by glucocorticoids, and TSH responsiveness to thyrotropin-releasing hormone (TRH) is frequently subnormal. Although basal levels are usually normal, the TSH concentration may be acutely low in patients treated with moderate- to high-dose glucocorticoids. Serum total thyroxine (T4) concentrations are usually low normal because of a decrease in thyroxine-binding globulin, but free T4 levels are normal. Total and free T3 (triiodothyronine) concentrations may be low, because glucocorticoid excess decreases the conversion of T4 to T3 and increases conversion to reverse T3. Despite these alterations, manifestations of hypothyroidism are not apparent.
Gonadal function—Glucocorticoids also affect gonadotropin and gonadal function. In males, they inhibit gonadotropin secretion, as evidenced by decreased responsiveness to administered gonadotropin-releasing hormone (GnRH) and subnormal plasma testosterone concentrations. In females, glucocorticoids also suppress luteinizing hormone (LH) responsiveness to GnRH, resulting in suppression of estrogens and progestins with inhibition of ovulation and amenorrhea.
- Peptic ulcer—The role of steroid excess in the production or reactivation of peptic ulcer disease is controversial. However, there appears to be a modest independent effect of glucocorticoids to promote peptic ulcer disease (relative risk about 1.4), and when this effect is combined with that of nonsteroidal anti-inflammatory drugs there is a synergistic interaction that considerably increases the risk.
- Ophthalmologic effects—Intraocular pressure varies with the level of circulating glucocorticoids and parallels the circadian variation of plasma cortisol levels. In addition, glucocorticoids in excess increase intraocular pressure in patients with open-angle glaucoma. Glucocorticoid therapy may also cause cataract formation. Central serous chorioretinopathy, an accumulation of subretinal detachment, may also complicate endogenous or exogenous glucocorticoid excess.
Adrenal Androgens
The direct biologic activity of the adrenal androgens (androstenedione, DHEA, and DHEA sulfate) is minimal, and they function primarily as precursors for peripheral conversion to the active androgenic hormones, testosterone and dihydrotestosterone. Thus, DHEA sulfate secreted by the adrenal androgens undergoes limited conversion to DHEA; this peripherally converted DHEA and that secreted by the adrenal cortex can be further converted in peripheral tissues to androstenedione, the immediate precursor of the active androgens.
The actions of testosterone and dihydrotestosterone are described in Chapter 12. This section will deal only with the adrenal contribution to androgenicity.
In males with normal gonadal function, the conversion of adrenal androstenedione to testosterone accounts for less than 5% of the production rate of this hormone, and thus the physiologic effect is negligible. In adult males, excessive adrenal androgen secretion has no clinical consequences; however, in boys, it causes premature penile enlargement and early development of secondary sexual characteristics.
In females, the adrenal substantially contributes to total androgen production by the peripheral conversion of androstenedione to testosterone. In the follicular phase of the menstrual cycle, adrenal precursors account for two-thirds of testosterone production and one-half of dihydrotestosterone production. During midcycle, the ovarian contribution increases, and the adrenal precursors account for only 40% of testosterone production.
In females, abnormal adrenal function as seen in Cushing syndrome, adrenal carcinoma, and congenital adrenal hyperplasia results in excessive secretion of adrenal androgens, and their peripheral conversion to testosterone results in androgen excess, manifested by acne, hirsutism, and virilization.
Laboratory Evaluation
Cortisol and the adrenal androgens are measured by specific plasma assays. Certain urinary assays, particularly measurement of 24-hour urine free cortisol, are also useful. In addition, plasma concentrations of ACTH can be determined. The assays for plasma steroids commonly used measure the total hormone concentrations and are therefore influenced by alterations in plasma-binding proteins. Furthermore, because ACTH and the plasma concentrations of the adrenal hormones fluctuate markedly (see Figure 9–7), single plasma measurements are frequently unreliable. Thus, plasma levels must be interpreted cautiously, and more specific diagnostic information is usually obtained by performing appropriate dynamic tests (stimulation and suppression) or other tests that reflect cortisol secretory rate.
Plasma ACTH measurements are extremely useful in the diagnosis of pituitary-adrenal dysfunction. The normal range for a plasma ACTH, using a sensitive immunoradiometric assay (IRMA) or immunochemiluminometric assay (ICMA), is 9 to 52 pg/mL (2-11.1 pmol/L).
Plasma ACTH levels are most useful in differentiating pituitary causes from adrenal causes of adrenal dysfunction as follows:
- (1) In adrenal insufficiency due to primary adrenal disease, plasma ACTH levels are elevated. Conversely, in pituitary ACTH deficiency and secondary hypoadrenalism, plasma ACTH levels are inappropriately normal or less than 10 pg/mL (2.2 pmol/L).
- (2) In Cushing syndrome due to primary glucocorticoid-secreting adrenal tumors, plasma ACTH is suppressed, and a level less than 5 pg/mL (1.1 pmol/L) is diagnostic. In patients with Cushing disease (pituitary ACTH hypersecretion), plasma ACTH levels are inappropriately normal or elevated.
- (3) Plasma ACTH levels are usually markedly elevated in the ectopic ACTH syndrome, but there is a considerable amount of overlap with levels seen in Cushing disease. In addition, values lower than expected may be observed rarely in ectopic ACTH syndrome when the two-site immunoradiometric assay (IRMA) is used; this assay may not detect high-molecular-weight precursors of ACTH that can be produced in ectopic ACTH syndrome.
- (4) Plasma ACTH levels are also elevated in patients with the common forms of congenital adrenal hyperplasia and are useful in the diagnosis and management of these disorders (see Chapters 10 and 14).
The most common methods of measurement of plasma cortisol are radioimmunoassay, enzyme-linked immunosorbent assay (ELISA), high-performance liquid chromatography (HPLC), and liquid chromatography tandem mass spectroscopy (LC/MS/MS). These methods measure total cortisol (both bound and free) in plasma. Most commonly used drugs and medications do not interfere with this assay. In contrast to radioimmunoassays, HPLC and LC/MS/MS do not demonstrate cross-reactivity with synthetic glucocorticoids.
The diagnostic utility of single plasma cortisol concentrations is limited by the episodic nature of cortisol secretion and its appropriate elevations during stress. As explained below, more information is obtained by dynamic testing of the HPA axis.
Normal values—Normal plasma cortisol levels vary with the method used and time of day the sample is obtained. With radioimmunoassay, levels at 8 am range from 3 to 20 μ g/dL (80-550 nmol/L) and average 10 to 12 μ g/dL (275.9-331.1 nmol/L). Values obtained later in the day are lower and at 4 pm are approximately half of morning values. At 10 pm to 2 am, the plasma cortisol concentrations by these methods are usually less than 3 μ g/dL (80 nmol/L).
Levels during stress—Cortisol secretion increases in patients who are acutely ill, during surgery, and following trauma. Plasma concentrations may reach greater than 40 to 60 μ g/dL (1100-1655 nmol/L).
High-estrogen states—The total plasma cortisol concentration is also elevated with increased CBG-binding capacity, which occurs most commonly when circulating estrogen levels are high (eg, during pregnancy and when exogenous estrogens or oral contraceptives are being used). In these situations, plasma cortisol may reach levels two to three times normal.
Other conditions—CBG levels may be increased or decreased in other situations, as discussed above in the sections on circulation and metabolism. Total plasma cortisol concentrations may also be increased in severe anxiety, endogenous depression, starvation, anorexia nervosa, alcoholism, and chronic kidney disease.
Cortisol in the saliva is in equilibrium with the free and biologically active cortisol in the blood. Salivary cortisol concentrations are not affected by changes in serum cortisol–binding proteins, by salivary flow or composition, and they are stable at room temperature for many days. Measurements of salivary cortisol can be obtained from late-night, ambulatory saliva samples, which are used as a means of establishing the presence or absence of Cushing syndrome. Salivary cortisol may also be used to obtain accurate free cortisol levels in patients with abnormal serum-binding proteins.
Saliva can easily be sampled at home by the patient using a variety of techniques, including the use of a commercially available sampling device or by passive drooling. The same methods that are employed to measure plasma cortisol, radioimmunoassay, ELISA, HPLC, and LC/MS/MS, are used to measure salivary cortisol concentrations.
Normal values—Reference ranges for late-night salivary cortisol concentrations are dependent on the assays employed, but with radioimmunoassay and ELISA, the normal salivary cortisol level at midnight is generally less than 0.15 μ g/dL (4 nmol/L).
Diagnostic utility—As with single plasma cortisol measurements, randomly obtained salivary cortisol concentrations are of limited use. However, plasma and salivary cortisol in normal individuals reach a nadir from 10 pm to 2 am. Patients with Cushing syndrome do not reach a normal nadir at this time, and several studies have shown that elevated late-night time salivary cortisol is a sensitive and specific diagnostic test for Cushing syndrome.