Despite significant advances in understanding of molecular pathobiology, the adoption of this knowledge and its application to drug development is sporadic. Consequently, the drug development process has remained intrinsically laborious and inefficient. Translational research seeks to improve this process by integrating scientific advances into well-defined clinical challenges and opportunities. The focus of this article is on cancer therapeutics, specifically, 2 biotechnology organizations’ advances in oncology: (1) optimizing the safety and efficacy of a novel DNA cross-linking small molecule, palifosfamide, and (2) bringing synthetic biology into clinical practice using a controllable gene therapy strategy with intratumorally injected interleukin-12 DNA, a potent cytokine.
Key points
- •
Translational research in biotech is predicated on cross-functional disciplines poised for identifying druggable targets and advancing the most promising ones through a gauntlet of scientific and regulatory hurdles.
- •
Synthetic biology uses synthetic DNA to express both exon-coded and intron-coded protein sequences to design, test, and build DNA-based therapeutics involving an engineering philosophy.
- •
Translational medicine holds the promise of accelerating the development of meaningful therapeutic drugs to address unmet medical needs. It starts with a practical question around a clinical issue rather than a fundamental biologic question, a “how” rather than a “why.”
The bench-to-bedside process
A New Era of Cancer Drug Development—Breaking Down the Walls: Academic, Government, and Industry Partnership
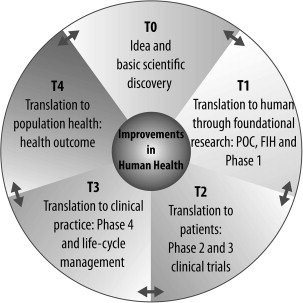
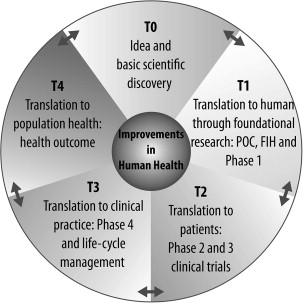
Translational medicine holds the promise of accelerating the development of meaningful therapeutic drugs to address unmet medical needs. It starts with a practical question around a clinical issue rather than a fundamental biologic question, a “how” rather than a “why.” The ultimate translational outcome may well be a new drug, but it can also be a combination of drugs, a device, a biomarker, or myriad other outcomes. Although the initial idea is often based on an unmet clinical need, the tools of basic research, including omics (eg, genomics or proteomics) and mechanistic experiments, serve to initiate validation of the idea in cell-based and pilot animal studies (T0) ( Figs. 1 and 2 ). Ideas that show promise for further development are then evaluated in T1 evaluation with in vivo proof-of-concept studies followed by first in human (FIH) and small phase 1 clinical studies to evaluate safety. Successful T1 leads to an expanded phase of clinical trials to investigate the efficacy in the given indication (T2). Often mechanism of action and surrogate biomarkers are also co-investigated and developed at this stage. Transition of this stage to a larger clinical evaluation permits large-scale evaluation of safety and efficacy (T3) (see Fig. 2 ). T4 steps of translational medicine involve transition to clinical practice and impact on large patient populations. This process is often iterative and learning from the clinical use of a drug informs the development of the next generation of novel and improved therapeutics (see Fig. 2 ).

The Biotech Advantage
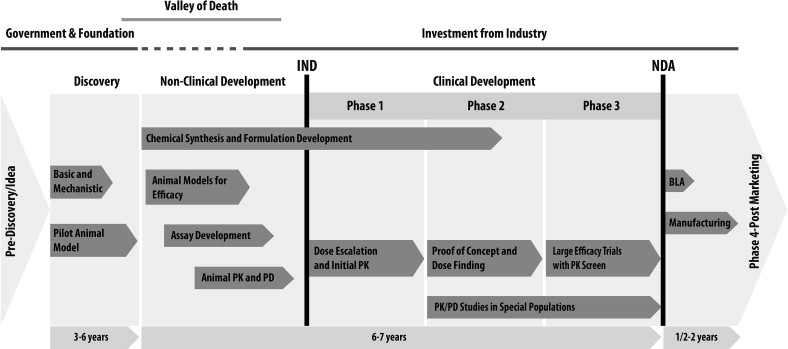
Translational research and drug development is a long and risky process (see Fig. 2 ). The advantage of having a major drug, however, that can benefit patients drives investments from government and the private sector. On average, the drug discovery process has taken approximately 15 years and costs at least $1 billion for each new drug approved. This process cannot be done in silos and requires an integrated and seamless collaboration between academia, industry, and governmental agencies (see Fig. 2 ; Fig. 3 ).
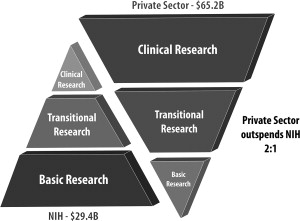
In the United States, academic institutions with funding from the National Institutes of Health (NIH) play a major role in the initial identification and characterization of basic early-stage T0 discoveries. Many promising basic discoveries, however, do not progress in the therapeutic development pipeline in academia because of the gap between early-stage bench research and implementation into clinical practice. This gap has been termed, the valley of death (see Fig. 2 ).
In many instances, this valley of death is a result of the traditional academic culture, with priorities of individual recognition (eg, publications and funding) and institutional net income through reimbursable activities. This culture promotes the creation of basic research silos and separate clinical silos that may not be compatible with teamwork and team recognition. Furthermore, successful and efficient drug development requires robust early decision making regarding the therapeutic potential of developmental candidates, which also contradicts the academic need for basic research and depth in mechanistic understanding. Additionally, the costs associated with lead discovery, optimization, and proof-of-concept nonclinical studies; restraints in NIH funding for translational research; and complexities of the regulatory path required for maintaining patient safety limit the translation of many discoveries to clinical practice at academic institutions.
The biotech industry provides a golden bridge from bench to bedside by providing the power of teamwork. Although pharmaceutical companies are also highly adept practitioners of bench-to-bedside and team-based translational research, it is generally believed that the smaller size of biotech organizations makes them more nimble, faster, and more efficient in dealing with bureaucratic challenges and rapidly changing environments. Biotech companies bring together highly qualified, intensely goal-oriented and collaborative teams of scientists and clinicians, regulatory staff, biostatisticians, data managers, research nurses, quality support, marketing, and management personnel to work together as a team and seamlessly advance a drug development candidate through a highly regulated environment that involves the Food and Drug Administration (FDA) and other global health authorities. Moreover, successful translation of promising therapeutics requires critical and strategic partnerships with physician-scientists at academic medical centers, who play critical roles in executing the FIH and phase 1 to phase 3 clinical trials and interacting with human subjects.
Legislations that Enhance Translational Research
In 1971, President Nixon declared the war on cancer and signed the National Cancer Act. Since then, there has been a significant increase in research to improve the understanding of cancer biology and the development of more efficacious therapies. This has resulted in considerable progress in translational research and in the treatment of certain cancer types, earliest and most significantly in childhood leukemia and testicular cancer. Cancer remains, however, a major cause of death. Several legislation statutes passed in the past 4 decades have addressed some of the limitations that are perceived to slow progress on the limited translation of promising drugs to clinical practice. For instance, due to widespread frustrations that discoveries made in academia were not being translated, the Bayh-Dole Act was put in place in 1980. This act obligates the academic institutions (that receive federal/NIH funding) to partner with industry to translate innovative ideas. Additionally, to encourage biotechnology and pharmaceutical companies to develop drugs for the treatment of rare diseases, the Orphan Drug Act of January 1983 was put in place. Moreover, in 1992, the Accelerated Approval Regulations were put in place to increase the speed of approval for novel or improved drugs that are intended to treat serious or life-threatening diseases. More recently, in 2004, the Critical Path Initiative started to improve the drug and device development processes and to enhance the quality of data generated during the translational research. As a consequence of all these measures, together with scientific and clinical advances, there was a record high of 39 new molecular entities approved by the FDA in 2012.
Investment in Translational Research in Biotech
Biotech is one of the only industries that can create potential value in the absence of immediate revenue generation. Investors, who are major stakeholders in biotechnology companies, are investing initially in ideas, human capital, proved and novel approaches, and innovative strategies for drug development. Investors ascribe value based on net present value. Biotech companies bring together the following pillars that are critical to the success of translational research.
Idea
Translational research typically starts with a patient-centered idea that could provide a solution to an unmet medical need. The scientific discoveries and innovations at the in silico , molecular, cellular, and in vivo levels that are made based on this idea can result in effective solutions when they are put into practice. Biotech companies often bring together internal scientists and external clinical key opinion leaders to evaluate and bring forward the most promising ideas for translation.
Human subjects
Human subjects are the heroes in translational research and are essential in making the transition to clinical practice possible by agreeing to participate in an experimental clinical study. They participate in all phases of drug development—from phase 1 to phase 3 and beyond. Generally phase 1 and phase 2 studies are considered within the domain of translational research. Although FIH studies in other areas of medicine are often performed in healthy volunteers, most anticancer drugs are initially tested in patients with cancers who have often failed earlier stages of therapy. In this way, in addition to pharmacodynamics (PD) and pharmacokinetic (PK) studies, investigators have an opportunity to assess early signals of clinical activity in patients and begin to make biologic, PK, and clinical correlations as soon as possible. Nevertheless, the key endpoint of FIH studies remains an assessment of the safety profile of the experimental drug and the establishment of the maximally tolerated dose that should be used in future studies. Patients willingly provide their consent to serve as test subjects for drugs with unknown side effects. In addition to accepting this risk, these patients understand that they may not be (and often are not) the direct beneficiaries of any resulting discoveries, therapeutic or financial. Their motivation is by and large altruistic. Without their active participation, the opportunities for innovation and clinical breakthroughs would come to a standstill.
Study principal investigators
The studies’ principal investigators (PIs) are often physician-scientists at academic medical centers who do not have any financial or personal conflicts of interest with the biotech company. PIs are personally responsible for conducting or supervising the conduct of the clinical research in human subjects and for protecting the rights, safety, and welfare of the subjects enrolled in the research. The PIs ensure that all in-human research is conducted in an ethical manner and in accordance with all federal, state, and local laws and regulations as well as institutional policies and that the subjects are fully informed of the research study and have clearly understood and then signed informed consent to participate in the study.
Nonclinical team
The main objectives of nonclinical studies for a candidate product are to identify and validate molecular targets for further clinical development, to understand the mechanism of action, and to determine the effect of the product on the body. This team performs cell-based and in vivo animal studies to determine what the most promising ideas for translation are. They also play an important role in understanding and defining nonclinical pharmacology-toxicology for both translation into the clinic and regulatory requirements. In addition, the members of this team carry out PD, PK, and pharmacology for absorption, distribution, metabolism, and excretion. Additionally, the nonclinical team performs studies to determine the mechanisms of action as well as the initial dose, schedule, and future dose increments that will be tested in humans. Potential dose-limiting and end-organ toxicities are identified and called to the attention of clinical investigators. PK studies in animal models that harbor tumors can yield valuable correlations between drug exposure (including the steady-state trough levels in the circulation and the area under the curve of the active drug) and safety or efficacy. These parameters can be readily monitored in subsequent FIH trials. Thus, the key goals of the nonclinical team are to delineate key toxicities in experimental models and anticipate and minimize harm to study subjects and to facilitate a more rational translational research plan.
Clinical operations and medical affairs
Clinical operations and medical affairs in many biotech and pharmaceutical companies are in charge of formulating the clinical development strategy for investigational drugs. Once an investigational drug has proved safe and efficacious in animal models and basic PK and PD parameters are established, it is necessary to translate these observations into the clinic. A medical team then engages in the designs of FIH and phase 1 clinical studies to identify a safe dose, the PK and PD of the investigational product, and the toxicity profile of the drug. Later stages of development involve the design of phase 2 and phase 3 clinical studies to identify initial signals of efficacy. The ultimate goal of this program is to obtain approval from the regulatory authorities to market the drug for the indication of choice. Depending on the disease targeted, these studies may involve comparison with drugs that have already been approved and constitute standard of care or with an inactive placebo.
Clinical operations
The clinical operations team is responsible for the successful execution of sponsored clinical studies in support of a regulatory filing (eg, NDA or Biologic License Application). The clinical team plays a key role in the design of study protocols, including, but not limited to, input on the schedule of assessments, determination of the number of sites necessary to support enrollment, and advice on which countries to include. They are in charge of making certain that study staff members are appropriately qualified and trained to execute the study protocol. The clinical team ensures that protocol-defined tests and samples are acquired correctly and at appropriate time points as well as that study data are accurate and consistent with the sites’ source documentation. Ultimately, clinical operations is accountable for (1) the quality of the study data, (2) overseeing the timely execution of study milestones, and (3) the management of the study budgets.
Chemistry, manufacturing, and control
The chemistry, manufacturing, and control (CMC) team is responsible for everything related to producing, analyzing, packaging, and delivering drugs for use in the clinical setting. The team evaluates physical and chemical properties of the drug to develop, optimize, and determine the proper scale for the manufacturing process. Whether it is a small or large molecule, the CMC team completes process development, chemical synthesis, and/or upstream processing for drug substance (eg, active pharmaceutical ingredient) manufacturing; formulation evaluation, dosage form development, and downstream purification are completed for drug product (eg, finished dosage form) manufacturing. The CMC team is also responsible for the development, qualification, and validation of all analytical methods used for process control or to analyze the drug for physical characteristics, potency, and purity at the time of release or during stability studies to establish retest/shelf-life dating.
Regulatory affairs
The regulatory function is absolutely vital in enabling the approval of safe and effective therapeutics so that they can become available worldwide. Regulatory affairs ensure regulatory compliance and prepare submissions and applications to agencies, such as the FDA, the European Medical Association, and other governmental agencies that oversee drug development ( Box 1 ). These agencies are more likely to approve new molecular entities if (1) an unmet medical need is being addressed; (2) strong data for evidence of efficacy are demonstrated statistically and clinically; (3) safety concerns are minimal and the benefits to subjects outweigh the risks; (4) current good clinical practices and current good manufacturing practices are followed; and (5) medical key opinion leaders support the approval of the new drug. These approvals come after heavy investment in preclinical development and clinical trials as well as a commitment to ongoing safety monitoring. Ultimately, it is all about the data, which must be assembled in a highly rigorous and regulated manner.
- •
IND: Based on nonclinical studies, a product is identified as a viable candidate for further development, and then the sponsor initiates the appropriate studies to establish that the product will not expose humans to unreasonable risks. The IND allows the sponsor to ship the study drug to clinical investigators at many sites.
- •
New Drug Application (NDA): NDA is the application for drug sponsors to formally apply to the FDA for approval of a new pharmaceutical for treatment of patients and sale and marketing in the United States. The data gathered during animal studies and human clinical trials under an IND become part of the NDA. The objectives of the NDA are to ensure that the drug is safe and efficacious for its intended use, its benefits outweigh its risk, the package insert labeling is appropriate, and the methods used for manufacturing maintain the drug’s identity, strength, quality, and purity.
- •
Biologic License Application: The Public Health Service Act requires the manufacturers of a biologic for sale to hold a license for that product. A Biologic License Application is submitted to the FDA and contains specific information on the chemistry, pharmacology (nonclinical and clinical), and manufacturing processes as well as the medical effects of the product. If the FDA requirements are met, the application is approved and a license is issued allowing marketing of the product.
Program management
Project managers often confront an overwhelming degree of complexity in extended, multifaceted projects, such as translational research and drug development. Program management leads cross-functional teams in all areas of drug development to advance therapies from early-stage development through commercial launch and life cycle management. Program managers lead without authority and manage through influence. They help to set metrics to ensure team responsibilities and that deadlines are met. They provide background information and context for issues and decisions. They collaborate with finance and line functions on budget activities as well as providing program visibility/transparency to senior management. In addition, they serve as a conduit for partnership communication and execution.
The bench-to-bedside process
A New Era of Cancer Drug Development—Breaking Down the Walls: Academic, Government, and Industry Partnership
Translational medicine holds the promise of accelerating the development of meaningful therapeutic drugs to address unmet medical needs. It starts with a practical question around a clinical issue rather than a fundamental biologic question, a “how” rather than a “why.” The ultimate translational outcome may well be a new drug, but it can also be a combination of drugs, a device, a biomarker, or myriad other outcomes. Although the initial idea is often based on an unmet clinical need, the tools of basic research, including omics (eg, genomics or proteomics) and mechanistic experiments, serve to initiate validation of the idea in cell-based and pilot animal studies (T0) ( Figs. 1 and 2 ). Ideas that show promise for further development are then evaluated in T1 evaluation with in vivo proof-of-concept studies followed by first in human (FIH) and small phase 1 clinical studies to evaluate safety. Successful T1 leads to an expanded phase of clinical trials to investigate the efficacy in the given indication (T2). Often mechanism of action and surrogate biomarkers are also co-investigated and developed at this stage. Transition of this stage to a larger clinical evaluation permits large-scale evaluation of safety and efficacy (T3) (see Fig. 2 ). T4 steps of translational medicine involve transition to clinical practice and impact on large patient populations. This process is often iterative and learning from the clinical use of a drug informs the development of the next generation of novel and improved therapeutics (see Fig. 2 ).
The Biotech Advantage
Translational research and drug development is a long and risky process (see Fig. 2 ). The advantage of having a major drug, however, that can benefit patients drives investments from government and the private sector. On average, the drug discovery process has taken approximately 15 years and costs at least $1 billion for each new drug approved. This process cannot be done in silos and requires an integrated and seamless collaboration between academia, industry, and governmental agencies (see Fig. 2 ; Fig. 3 ).
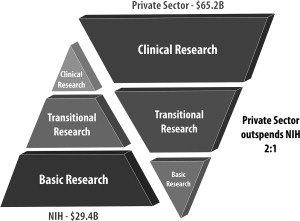
In the United States, academic institutions with funding from the National Institutes of Health (NIH) play a major role in the initial identification and characterization of basic early-stage T0 discoveries. Many promising basic discoveries, however, do not progress in the therapeutic development pipeline in academia because of the gap between early-stage bench research and implementation into clinical practice. This gap has been termed, the valley of death (see Fig. 2 ).
In many instances, this valley of death is a result of the traditional academic culture, with priorities of individual recognition (eg, publications and funding) and institutional net income through reimbursable activities. This culture promotes the creation of basic research silos and separate clinical silos that may not be compatible with teamwork and team recognition. Furthermore, successful and efficient drug development requires robust early decision making regarding the therapeutic potential of developmental candidates, which also contradicts the academic need for basic research and depth in mechanistic understanding. Additionally, the costs associated with lead discovery, optimization, and proof-of-concept nonclinical studies; restraints in NIH funding for translational research; and complexities of the regulatory path required for maintaining patient safety limit the translation of many discoveries to clinical practice at academic institutions.
The biotech industry provides a golden bridge from bench to bedside by providing the power of teamwork. Although pharmaceutical companies are also highly adept practitioners of bench-to-bedside and team-based translational research, it is generally believed that the smaller size of biotech organizations makes them more nimble, faster, and more efficient in dealing with bureaucratic challenges and rapidly changing environments. Biotech companies bring together highly qualified, intensely goal-oriented and collaborative teams of scientists and clinicians, regulatory staff, biostatisticians, data managers, research nurses, quality support, marketing, and management personnel to work together as a team and seamlessly advance a drug development candidate through a highly regulated environment that involves the Food and Drug Administration (FDA) and other global health authorities. Moreover, successful translation of promising therapeutics requires critical and strategic partnerships with physician-scientists at academic medical centers, who play critical roles in executing the FIH and phase 1 to phase 3 clinical trials and interacting with human subjects.
Legislations that Enhance Translational Research
In 1971, President Nixon declared the war on cancer and signed the National Cancer Act. Since then, there has been a significant increase in research to improve the understanding of cancer biology and the development of more efficacious therapies. This has resulted in considerable progress in translational research and in the treatment of certain cancer types, earliest and most significantly in childhood leukemia and testicular cancer. Cancer remains, however, a major cause of death. Several legislation statutes passed in the past 4 decades have addressed some of the limitations that are perceived to slow progress on the limited translation of promising drugs to clinical practice. For instance, due to widespread frustrations that discoveries made in academia were not being translated, the Bayh-Dole Act was put in place in 1980. This act obligates the academic institutions (that receive federal/NIH funding) to partner with industry to translate innovative ideas. Additionally, to encourage biotechnology and pharmaceutical companies to develop drugs for the treatment of rare diseases, the Orphan Drug Act of January 1983 was put in place. Moreover, in 1992, the Accelerated Approval Regulations were put in place to increase the speed of approval for novel or improved drugs that are intended to treat serious or life-threatening diseases. More recently, in 2004, the Critical Path Initiative started to improve the drug and device development processes and to enhance the quality of data generated during the translational research. As a consequence of all these measures, together with scientific and clinical advances, there was a record high of 39 new molecular entities approved by the FDA in 2012.
Investment in Translational Research in Biotech
Biotech is one of the only industries that can create potential value in the absence of immediate revenue generation. Investors, who are major stakeholders in biotechnology companies, are investing initially in ideas, human capital, proved and novel approaches, and innovative strategies for drug development. Investors ascribe value based on net present value. Biotech companies bring together the following pillars that are critical to the success of translational research.
Idea
Translational research typically starts with a patient-centered idea that could provide a solution to an unmet medical need. The scientific discoveries and innovations at the in silico , molecular, cellular, and in vivo levels that are made based on this idea can result in effective solutions when they are put into practice. Biotech companies often bring together internal scientists and external clinical key opinion leaders to evaluate and bring forward the most promising ideas for translation.
Human subjects
Human subjects are the heroes in translational research and are essential in making the transition to clinical practice possible by agreeing to participate in an experimental clinical study. They participate in all phases of drug development—from phase 1 to phase 3 and beyond. Generally phase 1 and phase 2 studies are considered within the domain of translational research. Although FIH studies in other areas of medicine are often performed in healthy volunteers, most anticancer drugs are initially tested in patients with cancers who have often failed earlier stages of therapy. In this way, in addition to pharmacodynamics (PD) and pharmacokinetic (PK) studies, investigators have an opportunity to assess early signals of clinical activity in patients and begin to make biologic, PK, and clinical correlations as soon as possible. Nevertheless, the key endpoint of FIH studies remains an assessment of the safety profile of the experimental drug and the establishment of the maximally tolerated dose that should be used in future studies. Patients willingly provide their consent to serve as test subjects for drugs with unknown side effects. In addition to accepting this risk, these patients understand that they may not be (and often are not) the direct beneficiaries of any resulting discoveries, therapeutic or financial. Their motivation is by and large altruistic. Without their active participation, the opportunities for innovation and clinical breakthroughs would come to a standstill.
Study principal investigators
The studies’ principal investigators (PIs) are often physician-scientists at academic medical centers who do not have any financial or personal conflicts of interest with the biotech company. PIs are personally responsible for conducting or supervising the conduct of the clinical research in human subjects and for protecting the rights, safety, and welfare of the subjects enrolled in the research. The PIs ensure that all in-human research is conducted in an ethical manner and in accordance with all federal, state, and local laws and regulations as well as institutional policies and that the subjects are fully informed of the research study and have clearly understood and then signed informed consent to participate in the study.
Nonclinical team
The main objectives of nonclinical studies for a candidate product are to identify and validate molecular targets for further clinical development, to understand the mechanism of action, and to determine the effect of the product on the body. This team performs cell-based and in vivo animal studies to determine what the most promising ideas for translation are. They also play an important role in understanding and defining nonclinical pharmacology-toxicology for both translation into the clinic and regulatory requirements. In addition, the members of this team carry out PD, PK, and pharmacology for absorption, distribution, metabolism, and excretion. Additionally, the nonclinical team performs studies to determine the mechanisms of action as well as the initial dose, schedule, and future dose increments that will be tested in humans. Potential dose-limiting and end-organ toxicities are identified and called to the attention of clinical investigators. PK studies in animal models that harbor tumors can yield valuable correlations between drug exposure (including the steady-state trough levels in the circulation and the area under the curve of the active drug) and safety or efficacy. These parameters can be readily monitored in subsequent FIH trials. Thus, the key goals of the nonclinical team are to delineate key toxicities in experimental models and anticipate and minimize harm to study subjects and to facilitate a more rational translational research plan.
Clinical operations and medical affairs
Clinical operations and medical affairs in many biotech and pharmaceutical companies are in charge of formulating the clinical development strategy for investigational drugs. Once an investigational drug has proved safe and efficacious in animal models and basic PK and PD parameters are established, it is necessary to translate these observations into the clinic. A medical team then engages in the designs of FIH and phase 1 clinical studies to identify a safe dose, the PK and PD of the investigational product, and the toxicity profile of the drug. Later stages of development involve the design of phase 2 and phase 3 clinical studies to identify initial signals of efficacy. The ultimate goal of this program is to obtain approval from the regulatory authorities to market the drug for the indication of choice. Depending on the disease targeted, these studies may involve comparison with drugs that have already been approved and constitute standard of care or with an inactive placebo.
Clinical operations
The clinical operations team is responsible for the successful execution of sponsored clinical studies in support of a regulatory filing (eg, NDA or Biologic License Application). The clinical team plays a key role in the design of study protocols, including, but not limited to, input on the schedule of assessments, determination of the number of sites necessary to support enrollment, and advice on which countries to include. They are in charge of making certain that study staff members are appropriately qualified and trained to execute the study protocol. The clinical team ensures that protocol-defined tests and samples are acquired correctly and at appropriate time points as well as that study data are accurate and consistent with the sites’ source documentation. Ultimately, clinical operations is accountable for (1) the quality of the study data, (2) overseeing the timely execution of study milestones, and (3) the management of the study budgets.
Chemistry, manufacturing, and control
The chemistry, manufacturing, and control (CMC) team is responsible for everything related to producing, analyzing, packaging, and delivering drugs for use in the clinical setting. The team evaluates physical and chemical properties of the drug to develop, optimize, and determine the proper scale for the manufacturing process. Whether it is a small or large molecule, the CMC team completes process development, chemical synthesis, and/or upstream processing for drug substance (eg, active pharmaceutical ingredient) manufacturing; formulation evaluation, dosage form development, and downstream purification are completed for drug product (eg, finished dosage form) manufacturing. The CMC team is also responsible for the development, qualification, and validation of all analytical methods used for process control or to analyze the drug for physical characteristics, potency, and purity at the time of release or during stability studies to establish retest/shelf-life dating.
Regulatory affairs
The regulatory function is absolutely vital in enabling the approval of safe and effective therapeutics so that they can become available worldwide. Regulatory affairs ensure regulatory compliance and prepare submissions and applications to agencies, such as the FDA, the European Medical Association, and other governmental agencies that oversee drug development ( Box 1 ). These agencies are more likely to approve new molecular entities if (1) an unmet medical need is being addressed; (2) strong data for evidence of efficacy are demonstrated statistically and clinically; (3) safety concerns are minimal and the benefits to subjects outweigh the risks; (4) current good clinical practices and current good manufacturing practices are followed; and (5) medical key opinion leaders support the approval of the new drug. These approvals come after heavy investment in preclinical development and clinical trials as well as a commitment to ongoing safety monitoring. Ultimately, it is all about the data, which must be assembled in a highly rigorous and regulated manner.
Related posts:
 Targeting Receptor Tyrosine Kinases in Solid Tumors
Targeting the NF-κB Pathway in Cancer Therapy
Targeting the p53 Pathway
Targeting Receptor Tyrosine Kinases in Solid Tumors
Targeting the NF-κB Pathway in Cancer Therapy
Targeting the p53 Pathway
 Advances in Molecular and Clinical Subtyping of Breast Cancer and Their Implications for Therapy
Biomarkers and Targeted Therapeutics in Colorectal Cancer
Translational Research in Endocrine Surgery
Advances in Molecular and Clinical Subtyping of Breast Cancer and Their Implications for Therapy
Biomarkers and Targeted Therapeutics in Colorectal Cancer
Translational Research in Endocrine Surgery
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


