Cancer is a complex disease capable of manifesting itself as many different disease phenotypes occurring in virtually any tissue of the body. This complexity can be the result of inherited single gene mutations in germline DNA, the accumulation of acquired genomic alterations over time, or the combination of both germline mutations and somatic genomic alterations.1 There are many examples of inherited or de novo mutations in the germline contributing to cancer susceptibility. These mutations include single nucleotide polymorphisms (SNPs), small insertions and deletions (InDels), as well as structural variants such as copy number variants, translocations, and inversions. In addition, somatic genetic and epigenetic changes are known to be involved in the earliest steps of cancer initiation within normal cells and during the proliferation of these transformed cells to form a precursor or in situ cancer.2 As transformed cells continue to grow, they undergo further genomic alterations taking on the invasive, immortal, and eventually metastatic properties common to most types of cancer. Mutations in specific and well-characterized genes, occurring during these earliest steps of tumor formation, appear to cause further genetic instability within the tumor cells resulting in the accumulation of even more deleterious genomic alterations, sometimes referred to as acquiring a “mutator phenotype.”3 The summation of these genetic, epigenetic, and genomic events provides the tumor cells with a distinct growth advantage characteristic of cancer and which can confer resistance to therapy. In addition, there remains an open question about the potential for viral genomes to interact with the human genome throughout life to initiate the molecular changes that can result in cancer.
Recent and rapid advances in the technology supporting molecular biologic and genomic studies and novel methods in bioinformatic analysis of large-scale data sets have substantially increased our knowledge of both normal and abnormal cell growth, especially as these phenotypic changes relate to the genome. Improvements in whole genome sequencing (WGS), whole exome sequencing (WES) through targeted exome capture, RNA sequencing for expression, bisulfate sequencing for DNA methylation as a mechanism of epigenetic gene modification, and the development of a wide variety of bioinformatic tools for the interpretation of genomic data have empowered new insights into classic cell biology and biochemistry of tumor cells.
As our knowledge of signaling pathways, genomic networks, intra- and extracellular communication, cell-cycle regulation, programmed cell death, and cell differentiation has progressed, we have come to understand the very heterogeneous nature of both the tumor and the surrounding host tissue in which it resides. We now know that the tumor is far less clonal than originally thought. Its genomic and cellular heterogeneity or multi-clonal nature is seen at the level of the tumor genome and this is manifested within the tumor by differing states of cellular differentiation and function. Even further, the genomic changes that characterize the primary tumor usually differ from those found in the tumor’s various metastatic sites.4 This tumor heterogeneity is also illustrated by a tumor cell subpopulation with unique surface markers termed “cancer stem or initiator cells” that maintain the power to self-renew, to have unlimited growth, and to undergo further differentiation. These stem-like cancer cells are several differentiation steps removed from the earlier stages of pluripotent stem cells, but retain the power of symmetrical cell division and are tumorigenic in contrast to the rest of the more differentiated cells within the tumor, which lack this self-renewal capacity.5
Equally important to the process of tumor progression is the presence of morphologically normal host cells of the tumor microenvironment. While their microscopic appearance is unchanged, these nontumorigenic host cells are genomically reprogrammed in terms of their expressed genes and resultant proteomic functions in order to communicate with and support the tumor’s growth, invasion, and spread.6
This new appreciation of a much higher order of complexity and cellular organization in malignancy, driven by disease-associated genomic alterations—including SNPs and InDels as well as structural variants—is expanding the field of new diagnostic, therapeutic, prognostic, and predictive biomarkers in cancer underscoring an appreciation for complete and sometimes prophylactic surgical resection. In the past, serial imaging was the key biomarker for tumor diagnosis, and tumor progression monitoring and evaluation of response to therapy. While anatomic and functional imaging technology has undergone tremendous advances and remain central in the care of the cancer patient, imaging often fails to accurately quantify actual changes in the patient’s viable tumor mass. As a result, there is a vital need for other highly predictive biomarkers of high sensitivity and specificity to provide the earliest possible diagnosis and most accurate indications of prognosis including precise real-time measurement of tumor cell burden during treatment. In addition to knowing where and how much of the tumor is present, it is critical to know the disease-causing potential of the tumor based on the genomic-driven cellular processes resulting in tumor growth and metastasis.
Biomarkers occur across the continuum of biologic changes associated with cancer. Serum measurement of tumor-specific antigens (CA 15-3, CA 19-9, CA 125, PSA, AFP, S100, etc.) has been useful as a gross indicator of tumor presence, but few of these markers have had the required sensitivity to stand alone as a fully informative biomarker. Since the “War on Cancer” started in 1971, many antibody-based biomarkers and therapies have been developed. Many cell-based biomarkers and immunotherapies using cytotoxic T cells, dendritic cells, natural killer cells, and lymphokine-activated killer cells have been developed with only one, Provenge (sipuleucel-T), currently approved and used for the treatment of prostate cancer.7 In addition to the more common immunological markers, some biomarkers detect the level of DNA, RNA, and blood-borne secreted proteins (EGFR, HER-2, KIT, BRAF, etc.). Relevant biomarkers can also include inherited genetic variants that account for somatic alterations (BCR-ABL, BRCA1/BRCA2, etc.) and predict increased risk of developing cancer or the presence of viruses (HPV, HIV, etc.) that may precede the onset of cancer. More recently, the detection of circulating DNA fragments with tumor-specific sequence variants holds promise as future cancer biomarkers, while the utility of detecting circulating tumor cells as a biomarker utilizing either genomic or immunologic assays appears to be waning.
This chapter reviews the relationship of the genome to cancer initiation and progression, with a focus on how these genomic alterations relate to the development of biomarkers for screening, diagnosis, prognosis, and therapeutic monitoring. As this is a surgical text, the emphasis will be on epithelial tumors.
Maintenance of a stable genome in somatic tissues through the processes of cell cycle regulation and apoptosis, and genomic and immunologic surveillance, and genomic editing mechanisms depends on cellular genomic makeup and the microenvironment of the cell. Maintaining genome stability is essential to minimizing the opportunity for cancers to initiate, while surveillance mechanisms remove or correct defective genes and genomes, thereby limiting tumor progression. In spite of a massively redundant system to ensure stability, genomic instability is a key mechanism for tumorigenesis. An imbalance in the complex control mechanisms that regulate genomic stability and immunologic surveillance increases genomic variability.8
Deregulation of cell growth, persistence of a “stem-like” self-renewing cell subpopulation, and loss of programmed cell death are hallmarks of cancer cells. The same characteristics are shared by the normal processes of frequent cell division and cell growth required to replace the differentiated cells of our tissues and organs.9 The process of frequent cell division for tissue self-renewal occurs within a microenvironment. Although in the vast majority of times cellular division within the microenvironment is under the control of normal cellular processes that maintain the integrity of the normal cell and tissue, cells may be exposed to an environment that could cause direct DNA damage resulting in a change in cell processes and organ function and initiation of the cancer cell. There are constant opportunities for the occurrence of genomic errors that escape detection and/or correction, resulting in the progression of tumor cell growth. Through WGS of both germline DNA and the DNA extracted from primary and metastatic tumors, scientists have identified individuals with specific genomic variants that alter the genes whose proteins are required to maintain genomic stability and genes whose DNA-binding proteins are the regulators of normal gene transcription and translation. As a consequence, a biomarker for cancer initiation and progression could represent a variety of genomic or nongenomic changes (immunologic, environmental exposures, infectious agents, etc.) characteristic of the cellular alterations associated with these processes.
It is important to maintain a mental vision of the complexity of this process if one is to optimize opportunities for biomarker development. Some of the earliest cytogenetic biomarkers focused on the ability to use light microscopy to observe structural alterations of the 23 tightly compacted chromosomes contained in the cell’s nucleus. It is interesting to note that chromosomes and individual genes are nonrandomly located within the cell nucleus.10,11 In fact, they occupy unique locations related to one another and to physical sites within the nucleus such as the nuclear envelope. A chromosome positioned within the nucleus may in fact determine whether the genes it carries are active or not. Misteli and colleagues have shown that changes in nuclear positioning are gene specific and not the result of genomic instability in cancer.11,12 The Misteli laboratory recently reported that in breast cancer tissues a single gene, HES5, was repositioned in all tested human breast cancer samples as compared to its location in noncancer cells.12–14 HES5 is a transcription factor that functions in the Notch signaling pathway by binding to the notch proteins, a set of four highly conserved transmembrane receptor proteins involved in cell proliferation and frequently implicated in cancer.14 HES5 and other genes found to undergo nuclear repositioning in breast cancer tissues may prove to be a useful early diagnostic biomarker to identify early cancer and precancerous changes in tissues.
In addition to a set location within the nucleus, each chromosome has a defined size and distinctive higher order structure. At the most basic level, chromosomes comprise two strands of DNA that twist around each other in a clockwise direction to form a double helix. The two strands carry a unique sequence of paired bases, the purines, adenine and guanine, and the pyrimidines, cytosine, and thymine. These two DNA strands wind around nuclear proteins called histones, which fold the DNA into structures of increasing complexity. The presence of a double-stranded DNA molecule is an important factor in gene function and DNA replication. Genes packaged into higher order structures are inactive and relatively stable. Active regions of DNA unwind to become biologically active and structurally accessible to change. DNA replication during cell division is a biologic function that requires coordination of a number of components and functions including enzymes that duplicate DNA as well as those that proofread and repair DNA.15 Genomic instability can occur during DNA replication through the process of introducing new DNA sequence variants into the genome and through structural rearrangement. Genomic variants, either inherited or somatically acquired, occurring in genes encoding proteins that recognize and/or respond to sites of DNA damage or DNA replication errors and additional genes encoding proteins that actually perform the repair are all potential acquired risk factors in cancer initiation. Each of these biologic processes and the genes involved can serve as biomarkers for the initiation and progression of cancer.
Like DNA replication, the process of gene transcription involves multiple genes and regulatory proteins. Each gene has a defined noncoding promoter sequence, and frequently contains sequences that act as tissue-specific enhancers, usually upstream of the 5′ start site for the gene to be transcribed. Transcription is initiated by a complex of small proteins that come together to form a DNA-binding complex. As many as 60 to 70 proteins are involved in gene transcription. The protein coding sequence of the gene contains segments of noncoding bases, termed introns, which are located between the coding sequence, that is, exons. After the process of transcription to messenger RNA (mRNA), these intronic sequences are excised in a very precise manner to generate the continuous coding sequence, the exome, for subsequent steps of translation into protein. Errors in the precise excision of the introns, resulting even in the addition or deletion of a single nucleotide at the splice junction, could have a deleterious effect on protein formation. The processes of transcription and translation are extremely robust and dynamic, with proteins binding to DNA and RNA in a continuous flow of molecular activity. The process is far less compartmentalized and isolated than represented by most models. The potential, therefore, exists for the formation of RNA with sequence variations and different isoforms of the protein from the gene with absent or altered functional properties. To add to the complexity of the process of going from DNA to RNA to protein, it is relatively common for exons to be skipped, resulting in different normal proteins being made from the same gene. The number of these splice variant proteins formed from a given gene’s RNA has a special implication for genes relevant to cancer, and once again provides a window into the extreme complexity that can be operative in the initiation and progression of the disease. To make things even more complex, there is the potential loss of normal protein to protein communication and pathways that, for example, regulate cell-cycle switches or that result in alterations of the target receptors of those proteins. In addition, this is an opportunity for posttranslation disease causing changes such as those affecting protein phosphorylation and ubiquitylation. Each of the genes, control points, proteins, protein modifications, and protein interactions involved in this process represent potential biomarkers for the process of tumorigenesis.
Finally, there are three methods of epigenetic regulation of gene transcription that can impact the initiation and progression of cancer. These methods of regulation include DNA methylation, gene regulation by microRNA (miRNA), and the accessibility of chromatin DNA to binding by transcription factors.
The enzyme-driven methylation of DNA around CpG residues and longer CpG islands of DNA blocks gene transcription. The mechanisms and signals for DNA methylation and the rules that determine when methylated DNA is unmethylated and available for transcription are not completely understood. Blocking of transcription happens at varying levels within the genome, locally and regionally. In certain cases, it has been shown that tumor suppressor genes can be inactivated by methylation. An example of an epigenetic biomarker for cancer is the observation that in glioblastomas the combination of hypermethylation in the presence of certain IDH1 variants has significantly different clinical outcomes.16
Another example of epigenetic control of gene expression involves miRNAs which highly regulate the translation of mRNA transcripts into proteins.17 MiRNAs are small 19 to 24 nt sequences of RNA derived from noncoding regions of DNA and each miRNA can target multiple (100s) mRNAs. Over 1000 miRNAs have been described. They are generated from specific DNA sequences by successive cleavage by Drosha and Dicer nucleases.18 Their precursor transcripts contain hairpin formations. MiRNAs have generally been shown to target a specific binding sequence within the 3′UTR region of the mRNA but more recently there has been evidence that the miRNAs can target specific sequence sites within the actual coding region.
In cancer, when miRNA expression is elevated, the corresponding target genes have their expression significantly downregulated. In contrast, when the miRNA is downregulated, the target gene expression is upregulated. Several miRNAs have been implicated in different stages of tumorigenesis. For example, downregulation of miR-148a has been reported as a biomarker of the metastatic phenotype for a number of cancers.19 A relationship between inhibition of miR-148a expression and tumor metastasis was reported in a study showing silencing of miR-148a by gene methylation in several metastatic lesions of different tumor types.20 MiR-148a was also shown to be downregulated in the cancer-activated fibroblasts of human endometrial cancer.6 In this instance, WNT10B mRNA was shown to be the direct target of miR-148a and was, as a result, significantly upregulated in its expression and ability to stimulate endometrial cancer cells.6
There is an impressive degree of diversity in miRNA expression across different tumor types and the cells of their microenvironments. Thus, profiling of miRNA expression patterns may evolve to be an important diagnostic tool in classifying cancer subtypes and for predicting their clinical behavior. These miRNA expression patterns may prove to be of even greater biomarker value than signatures of protein-coding genes.
The third regulator of gene transcription involves the actual unwinding of the tightly condensed heterochromatin formation of the DNA. This unwinding provides access to the promoter enhancer region of the gene’s DNA in order for the transcription factors to complex and bind to the DNA to initiate gene transcription. Many proteins are involved in the process and as a result variants in the genes that encode these proteins may play a role in cancer risk. There are two types of chromatin, heterochromatin, and euchromatin. These strands of chromatin, not naked DNA, are the actual substrate for transcription. As noted, heterochromatin is densely packed, contains thicker fibers of chromatin, and much of its DNA consists of sequence repeats. Thus, heterochromatin is inaccessible to transcription factors and genetically silent. It does play a critical role, however, in maintaining structural integrity of key regions of the chromosomes such as the centromeres and telomeres.21 Loss of heterochromatin function has been observed to be associated with an increased cancer.
Transcription of genes occurs where there is more open and therefore accessible euchromatin. As expected, there exists a broad array of chromatin modifier proteins and chromatin readers. Modifiers include proteins that transfer or remove acetyl or methyl modifications from histones, as well as factors that move nucleosomes along the DNA, or remove the nucleosome from the DNA, and modifier proteins that change the histone composition of the nucleosomes. All of these hundreds of proteins are of course susceptible to inherited or acquired mutations that alter or ablate their proper function. In recent years many of these have been shown to be involved in cancer formation.
It is clear that many hundreds of genes involved in the various biochemical pathways take part in normal cellular metabolism, direct regulation of transcription, and regulation of the several epigenetic mechanisms. All of these genes, along with the RNAs and proteins generated from them, and any byproduct, metabolite, or cofactor associated with them, are potential biomarkers.
As we have already discussed, there are many ways that genetic variants causing loss of function or amplification of function in genes involved in normal cellular processes can contribute to the development of cancer. Dysregulation by sequence or structural variation of non-cancer-related genes can lead not only to initiation of cancer, but also to the metastatic progression of the malignancy. Proto-oncogenes, a class of genes responsible for the normal regulation of cell growth, can be hijacked by a mutation to function in ways that are deleterious to normal cellular processes. Mutations can negatively impact gene activity in proto-oncogenes, resulting in their conversion to growth-promoting oncogenes that act as drivers of neoplastic cell proliferation. Point mutations in “driver genes” are a highly sought after category of biomarker. They are relatively easy to identify and, as mutations in driver genes, represent the ability to detect tumors in the very early stages of development. In addition to point mutations, chromosomal translocations can occur and as such affect the position of the proto-oncogene, which in the case of the MYC proto-oncogene encodes a transcription factor that binds to and activates the transcription of other genes. A translocation affecting the MYC gene can convert it to an oncogene, leading to the development of Burkitt’s lymphoma.22 In these translocations, the MYC gene, located on the long arm of chromosome 8—in cytogenetic terms, the normal location of the MYC gene is referred to as 8q24—becomes abnormally activated when translocated near specific gene sequences (the most common of which is IGH, the immunoglobulin heavy chain locus on chromosome 14q32). This translocation results in loss of regulation of c-MYC expression converting the gene to a “driver oncogene.”23 Through processes such as increased gene amplification due to translocation or increased copy number of the gene, this overactivation leads to cell growth and proliferation.
Effective biomarkers for translocations fall into three classes: those that detect a change in copy number, those that detect a hybrid DNA molecule that spans a recombination event, and those that detect increases in the activity of the gene product (RNA or protein). Typically, these are different than the sequence variant biomarkers that have been discussed thus far. Assays for these biomarkers must be able to detect the consequences of rearrangement, such as a probe that contains adjacent sequences from two genes that may have undergone rearrangement or are designed to identify multicopies of the gene (copy number variants).
Another extensively characterized translocation example involves the “Philadelphia chromosome,” a truncated chromosome 22. The classic Philadelphia chromosome results from a reciprocal translocation between chromosomes 9 and 22 that converts the proto-oncogene ABL1 to an oncogene with greatly increased tyrosine kinase activity, most commonly associated with chronic myelogenous leukemia (CML).24 The translocation is also present in approximately 25% of adult ALL, 10% of pediatric ALL, and occasionally (but rarely) in AML. A molecularly based treatment for CML that acts by inhibiting the oncogene’s tyrosine kinase activity has been effective.25,26
Mutations that potentiate cancerous processes can also occur in tumor suppressor genes. Mutations in tumor suppressor genes can result in aberrant control of cell growth (these are termed gatekeeper tumor suppressor genes) or in further loss of genomic integrity (termed caretaker tumor suppressor genes). For example, heterozygously inherited mutations in RB1 can result in retinoblastoma, an ocular neoplasm. In individuals who inherit a mutation, retinoblastoma occurs when a second, somatic mutation affects the other, normal copy of the RB1 gene.27 Because both copies of the tumor suppressor gene no longer function appropriately—referred to as the “two-hit hypothesis” or “Knudson’s hypothesis” of cancer development—the result is abnormal regulation of proliferating cells in the cell cycle and of differentiated cells exiting from the cell cycle.28
On an individual/family level, both copies of the RB1 gene must be inactivated by mutations (again, the most common pattern is that one mutation is inherited while the other is somatic in origin) for clinical disease and the biomarker assay must be able to distinguish between the presence of one versus two copies of the gene carrying a mutation. The mechanism is reminiscent of a recessive condition, where both copies of a gene must be abnormal, though the inheritance pattern appears dominant when viewing a pedigree (Fig. 8-1). Unsurprisingly, given the normal function of the RB1 gene product, individuals with mutations in RB1 are prone to other types of cancer in addition to retinoblastoma; these may also involve somatic mutations in the affected tissue.29 The primary mutations described above, which at first occur in the initial transformed cell, allow other mutations to accrue. These other mutations result in additional loss of genomic integrity, and further contribute to tumor growth, spread, and tumor cell survival. Thus, in any one neoplasia, multiple mutations of diverse types may contribute to cancer development (Fig. 8-2).
FIGURE 8-1
Retinoblastoma pedigree. Hypothetical pedigree showing inheritance of retinoblastoma, due to mutations in RB1. Shaded symbols indicate clinically affected individuals. Though the inheritance pattern appears to be (autosomal) dominant, individuals will not be affected unless they both inherit a germline mutation (Mut, for Mutation) in RB1 as well as have a secondary, somatic mutation affecting the other normal (Nl, for Normal) copy (allele) of RB1. The individual with the concentric circles has not shown phenotypic signs of having the mutation, as the “second hit” has not yet occurred, but she is at high risk of disease.
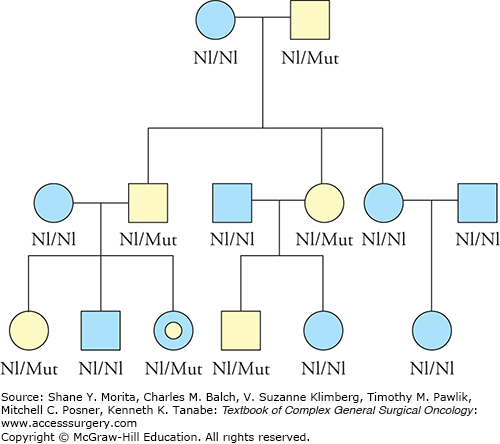
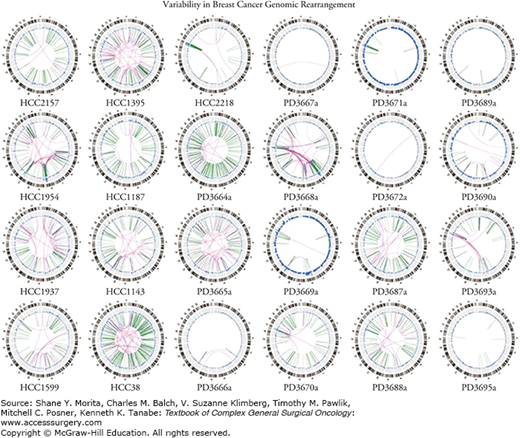
FIGURE 8-2
Variability in breast cancer genomic rearrangement. An example of the genomic heterogeneity detected in breast cancer specimens obtained from a series of patients. (Reproduced with permission from Stephens PJ, McBride DJ, Lin ML, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. December 24, 2009;462(7276):1005–1010.)
Though there are many millions of individual genetic variants that can be involved in oncologic disease, the functional alterations that contribute to cancer development can be defined by the following categories: self-sufficiency in growth signals; insensitivity to anti-growth signals; evasion of apoptosis (programmed cell death); limitless potential for replication; sustained angiogenesis; tissue invasion and metastasis, immunologic tolerance, and the ability to continuously self-renew.30–32 High-throughput technologies, such as tracking the transcriptome of tumor cells, can be used to follow and analyze tumor development, potentially revealing clinically relevant biomarkers related to each of the above categories, which can be used to develop algorithms to help guide therapy based on the molecular properties of an individual cancer.32,33 Given that there may be millions of genetic variants that are potentially involved in cancer and an equal number of scenarios by which genes without mutations may result in changes in the quantity or quality of a cancer-inducing protein, either through transcription or translation, it is important to recognize that the type of biomarker and the technology used to detect the biomarker are extremely variable.
Traditionally, discussions regarding medically significant genetic variants in malignancy have focused on changes within the genes themselves. However, as mentioned, there are many additional ways that genetic variants may contribute to cancer. First, in addition to a change within a gene, a genetic variant outside the actual coding exon sequence of a gene may alter the gene’s function. For example, a genetic variant may affect a regulatory region of a tumor suppressor gene. There is increasing attention being paid to these regulatory regions, and current evidence points to the fact that much of what was previously thought to be “junk DNA” left over as a relic of evolution may in fact play a critical function, though teasing out the exact biologic role of any given region is a formidable task.34 As described above, in addition to traditional genes that encode proteins, genes may encode other functional units, such as miRNAs. Genetic alterations affecting these miRNAs can act similarly to oncogenes or tumor suppressors.35 Second, it is easy to forget that it is proteins and functional RNA molecules that are the biologic machinery that carry out the functions of transcription, translation, posttranslational modification, protein transport systems, etc. Sequence variation(s) in genes that code for the molecules that perform these critical processes can result in differences in the individual protein’s affinity for its targets and thus potentially be a causative factor for disease. A comprehensive list of targets to monitor all theoretically possible cancer-related processes within the cell would have to include an unreasonably large number of assays for the many genes and processes. Although many potential targets can be assayed, it is only those that occur in a statistically significant number of tumors that make sense to develop into biomarkers for clinical application.
Alterations affecting a person’s genetic function may not cause direct DNA-level changes, or be easily detectable by interrogating the DNA sequence, thus necessitating attention to biomarkers such as those involving gene regulation or RNA or protein expression. For example, epigenetic alterations, such as changes affecting the methylation of tumor suppressor genes, may inappropriately activate or inactivate genes, resulting in or contributing to neoplastic processes.36 In reality, the oncologic process may occur due to the combinatorial effects of many genetic and other influences, and understanding this network, as well as using this knowledge to guide clinical care, provides many challenges yet strong potential.
Oncologic processes may involve “constitutional” genetic variants, or genetic changes that are found in all the different somatic cells in a person. Typically, this is because these genetic variants are inherited. Even on this most basic level, there are exceptions, as de novo genetic changes may occur at the zygote stage and may go on to be found in all of a person’s cells. Constitutional genetic variants may be silent while others contribute to a variety of biologic processes, some of which have health implications. Certain constitutional genetic variants, depending on the type of change and the involved gene or genetic regulatory region, may affect a person’s chance of developing cancer. A well-known example of this is the BRCA1 gene. In a model that requires two mutated genes to promote tumor formation, if one is constitutionally absent, it then may only take one mutation in the other copy of the BRCA1 gene to initiate cancer. The Sanger Cancer Gene Census (http://www.sanger.ac.uk/research/projects/cancergenome/census.html) contains a list of genes that scientific evidence suggests play a role in cancer. Though genetic variants are traditionally thought of as increasing a person’s risk of cancer, the converse may hold true as well; some genetic variants may result in decreased cancer risk.
Some human diseases, such as those related to inherited cancer predisposition syndromes, are best known to involve constitutional changes, while others, such as traditional “sporadic cancer,” are more classically thought of as involving somatic mutations. However, a given gene may be involved in a number of medical conditions, depending on the timing of the genetic variant, as well as properties specific to the type of variant. Proteus syndrome, a rare but dramatic overgrowth syndrome that involves oncologic manifestations, can be due to somatic mutations in the gene AKT1. The discovery of the cause of the Proteus syndrome was found by comparing tumor to normal tissue through exome sequencing.37 Germline mutations in the same gene can cause Cowden and Cowden-like syndromes, multiple hamartoma syndromes that involve a high risk of multiple cancer types including breast and thyroid cancer.38 Finally, somatic AKT1 mutations may also be part of the landscape of genetic alterations involved in other cancer processes, such as breast and endometrial cancer and meningiomas.39–41
It is important to note that not all individuals carry the same risk of cancer based on detectable constitutional sequence variants. There is a strong ancestral component to the inheritance of constitutional variants that range from founder effect scenarios, where a sequence variant in a relatively secluded population can trace that variant to a single individual, to a difference in minor allele frequencies for a given variant that differs between ethnicities. These differences can impact the incidence of disease in a population and the manner in which a family is counseled with regard to their risk.
Distinct from constitutional genetic variants, oncologic processes frequently also involve somatic genetic alterations that occur through imperfect replication and cell division. With somatic variants, unlike constitutional variants, the genetic change occurs after conception. That is, the somatic variant initially occurs in a single cell in a certain tissue, but is not present in all other cells that comprise a person. These somatic variants are also not present in the germ cells—the sperm and egg—and so cannot be passed down to the next generation. Somatic variants will then be passed on to daughter cells arising from the cell with the original somatic mutation. Many somatic variants will not have a measurable effect on cellular function, but randomly, some may affect certain genes in ways (described above) that can contribute to neoplastic processes. Perhaps what is most interesting is the knowledge that even when germline mutations in a specific oncogene or tumor suppressor gene exist, the vast majority of cells in an individual with such strong cancer risks never develops into cancer or even develops the changes associated with benign tumors. This must be assumed as strong evidence that cancer is most commonly the result of an accumulation of multiple genomic defects in the ultimately transformed cell.
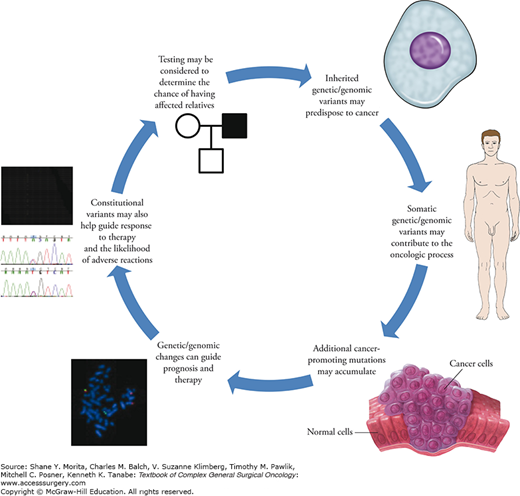
In addition to the role of genetic changes in causing and contributing to the overall oncologic mechanism, changes affecting the genetic material can frequently be used to investigate key clinical questions relating to diagnosis, prognosis, disease recurrence, and management (Fig. 8-3). These types of clinically oriented investigations often focus on cancer-related biomarkers (see Table 8-1 for a glossary of relevant terms).
FIGURE 8-3
Genetics/genomics circle. Genetic and genomic variants may contribute to the development of cancer, and may be relevant to a variety of clinical considerations. From top right: Constitutional variants, which are frequently inherited, may contribute to a person’s cancer risk; additional somatic variants (as well as other factors, such as environmental influences) may affect the development of cancer; once the oncologic process is initiated, multiple additional genetic/genomic variants that further contribute to neoplasia and malignancy may accrue; after identification, specific genetic and genomic factors of the neoplasia may be important in considering issues such as prognosis and optimal treatment modalities; additionally, “host” genetic factors not primarily related to cancer may be involved in prognosis, the efficacy of treatment, and further clinical parameters; in some instances, the presence of inherited genetic mutations may be queried in order to provide risk information for other family members.
Glossary of Relevant Terms (as applies to human genetics and genomics)
| Term | Definition |
|---|---|
| Allele | A version of a gene |
| Allelic heterogeneity | A condition or trait that may be caused by different mutations within the same gene |
| Biomarker | A biologic molecule that can be found in blood, tissue, or other body fluids, and which can be measured to correlate with the absence or presence of a medical condition or disease, as well as properties related to that disease, including therapeutic considerations |
| Caretaker tumor suppressor gene | A tumor suppressor gene that normally functions to maintain genomic integrity |
| Compound heterozygous | The presence of two different abnormal (mutant) alleles at a given locus |
| Constitutional genetic variant | A genetic variant found in all the different cells in a person (as opposed to a somatic variant) |
| Epigenetics | A change in gene regulation that occurs without altering the DNA-level genetic structure itself |
| Exome | All exons in a person (or cell) |
| Exon | The portion of a gene that ultimately encodes proteins |
| Expressivity | The nature and severity of a condition (usually described in relation to a penetrant mutation) |
| Gatekeeper tumor suppressor gene | A tumor suppressor gene that functions to maintain normal cell division and growth |
| Genetic variant | A DNA-level change that may or may not have a functional effect or result in pathogenicity |
| Genome | A person’s (or cell’s) entire genetic makeup |
| Heterozygous | The presence of two different alleles at a given locus (frequently, this refers to one mutation and one normal allele) |
| Homozygous | The presence of the same allele at a locus (this is frequently used to refer to two copies of the same mutation, but may also refer to homozygosity for the normal allele) |
| Locus heterogeneity | A condition that may be caused by mutations in a number of different possible genes |
| Mutation | A DNA-level change that results in a functional effect and causes pathogenicity/disease |
| Oncogene | A mutant version of a gene, which contributes to cancer development |
| Penetrance | The proportion of individuals with a given mutation who display a certain condition because of that mutation |
| Pharmacogenetics/pharmacogenomics | The application of genetics/genomics in the practice of drug development, testing, and use |
| Proband | The person (usually within a family) who first comes to medical attention because of a particular condition |
| Proto-oncogene | A gene that may be converted to an oncogene, which then contributes to cancer development |
| Somatic genetic variant | A genetic variant that occurred after conception, and which is present only in certain cells derived from the cell in which the somatic variant occurred (as opposed to a constitutional genetic variant) |
| Transcriptome | The set of RNA molecules produced by one or many cells |
| Tumor suppressor gene | A gene whose normal function involves suppression of oncologic processes by restraining cell division and growth, and maintaining genomic integrity |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


