Fig. 2.1
BRCA, DNA repair and the cell cycle (Foulkes and Shuen [9]). In response to DNA damage, BRCA1 mediates HR (depicted in the outer ring) and cell cycle regulation (depicted in the inner ring) when bound to different various macrocomplexes. Following a double-strand break, ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3-related) phosphorylate a number of downstream effectors, including H2AX, MRN (MRE11-RAD50-NBN), BRCA1 and its binding partner BARD1 (BRCA1-associated RING domain protein 1), initiating the DNA damage response (DDR). BRCA1 binds to BRIP1 (BRCA1-interacting protein 1), and SWI/SNF regulates histone deacetylases to open up the chromatin, perhaps allowing access of repair enzymes to the site of DNA damage. Following complex enzymatic modifications by ubiquitin and SUMO (small ubiquitin-like modifier), RAP80 (receptor-associated protein 80) and FAM175A (Abraxas) recruit BRCA1 and other downstream repair enzymes to the site of DNA damage. BRCA1, coupled with MRN and CtIP (C-terminal binding protein interacting protein), is involved in resecting the DNA ends to create single-stranded DNA (ssDNA), which is protected by RPA (replication protein A). The BRCA1/PALB2/BRCA2 macrocomplex is then required for RPA displacement and RAD51 loading onto ssDNA. Finally, RAD51 mediates sister chromatid strand invasion and homologous repair. Acting in parallel with the DNA damage response are BRCA1 complexes that regulate the cell cycle. BRCA1 coupled with BRIP1 and TOPBP1 regulates G1 – S and intra-S phase checkpoints, while BRCA1/MRN/CtIP and BRCA1/RAP80/FAM175A (Abraxas) regulate the G2 – M phase checkpoint
The involvement of BRCA1 and BRCA2 in DNA damage response (DDR) has led to the hypothesis that cells with a BRCA1 or BRCA2 defect could be more sensitive to alkylating agents that considerably increase DSBs and to molecules inhibiting DNA repair pathways other than HR, such as base excision repair (BER) [11]. Poly-ADP-ribose-polymerase inhibitors are an emerging family of DDR inhibitors (see Chap. 13 J Ledermann).
As mentioned above, BRCA1 and BRCA2 mutations cannot account for all severe breast/ovarian cancer families, suggesting that other predisposing genes have yet to be identified. Consequently, partner genes of BRCA1 and BRCA2, especially those involved in the HR pathway, are good candidates to be associated with high cancer risks and higher tumour sensitivity to alkylating agents or DDR inhibitors in case of gene inactivation. Numerous genes have been tested in association studies, for example, CHEK2 and CHEK1 involved in cell cycle control; ATM, MRE11A and NBN involved in the detection of DNA damage; and PALB2 and BRIP1 which are also Fanconi disease genes. Of note, no monoallelic deleterious mutation of RAD51 has yet been reported, probably because such mutations would be lethal. In contrast, mutations of RAD51 paralogs (duplicated genes during species evolution that have slightly diverged but still have very similar functions to those of the original gene) have been reported and are associated with an increased risk of ovarian cancer (see below). Most of these genes are currently at the stage of research, and genetic testing has not been proposed to date.
Histology of Ovarian Carcinoma in BRCA1/BRCA2 Mutation Carriers
Lakhani et al. compared the pathological characteristics of 178 BRCA1 and 29 BRCA2 OCs to those of 235 age-matched controls [12]. Both BRCA1 and BRCA2 tumours were of higher grade than control tumours (p <0.0001 and p = 0.028, respectively). Well-differentiated and grade 1 tumours do exist in BRCA1/BRCA2 mutation carriers but tend to be rare. Similarly to sporadic cases, papillary serous OC is the most prevalent type, observed in 44 and 48 % of BRCA1 and BRCA2 mutation carriers, respectively, followed by the endometrioid type, 36 and 38 % in BRCA1 and BRCA2 mutation carriers, respectively. The frequency of serous tumours is reported to be significantly higher among BRCA1 mutation carriers (OR 1.84, 95 %CI 1.21–2.79), while the frequency of mucinous tumours is much lower (OR 0.13, 95 % CI 0.05–0.34, p <0.0001). The distribution of histological types in BRCA2 tumours is similar to that in BRCA1 tumours but not significantly different from the control distribution. The frequency of borderline tumours does not appear to be increased in BRCA1/BRCA2 mutation carriers. In the study by Zhang et al., no BRCA1/BRCA2 mutation was identified in a series of 112 cases of unselected mucinous carcinomas [13]. As in the general population, clear cell forms and carcinosarcomas are rare. In summary, BRCA1/BRCA2 OCs are classically poorly differentiated and of high grade, corresponding to the “type 2” pathway of ovarian carcinogenesis [14]. OCs in BRCA1/BRCA2 mutation carriers are thought to arise from serous intraepithelial tubal carcinoma (STIC) in the fallopian tubes, associated with TP53 somatic mutations [15].
Prevalence of BRCA1/BRCA2 and Other HR Pathway Gene Germline Mutations Among Ovarian Cancer Cases
Before reporting the prevalence of BRCA1/BRCA2 germline mutations in women with OC, it is useful to recall the prevalence of these mutations in the general population (males and females). According to the Anglian Breast Cancer Study, and taking into account the Hardy and Weinberg law, the allelic frequency for BRCA1 mutations in the general population was estimated to be 0.051 % (95 % CI: 0.021 – 0.125 %), and the allelic frequency for BRCA2 mutations was estimated to be 0.068 % (95 % CI: 0.033 – 0.141 %). The frequencies of BRCA1 and BRCA2 mutation carriers were therefore estimated to be 1/974 and 1/734, respectively. In other words, the frequency of BRCA1/BRCA2 mutation carriers in the general population is about 1/400 [16]. In the study by Song et al. described below, the observed frequency of BRCA1/BRCA2 mutation carriers among the 1528 cancer-free controls was 0.37 % (one BRCA1 mutation, 4 BRCA2 mutations), corresponding to 1/270 BRCA1/BRCA2 carriers in the general population, with 1/1428 BRCA1 mutation carrier and 1/333 BRCA2 mutation carrier [17].
Numerous studies have examined the prevalence of BRCA1/BRCA2 germline deleterious mutations in women with OC. The most recent studies performed in the largest series were based on a molecular testing approach that was as complete as possible [17, 18].
Song et al. performed germline analyses of BRCA1, BRCA2, MLH1, MSH2, MSH6 and PMS2 genes in a series of 2222 women with invasive OC unselected for breast or ovarian cancer and in 1528 controls. Proportions of histological subtypes, serous (57 %), endometrioid (14 %), clear cell (8.6 %), mucinous (7.1 %) and high grade (66 %), were consistent with unselected OCs. Among the 2222 OCs, 178 (8 %) BRCA1/BRCA2 mutation carriers were identified: 84 BRCA1 mutation carriers (3.8 %) and 94 BRCA2 mutation carriers (4.2 %). The proportion of BRCA1/BRCA2 carriers was higher in the high-grade subgroup, with 11 % of carriers.
Alsop’s study, conducted in a series of 1001 consecutive cases of non-mucinous, non-borderline OC, identified 141 BRCA1/BRCA2 mutation carriers: 14.1 % (95 % CI: 11.9–16.3). About 2/3 of these cases were BRCA1 mutation carriers (88 cases), and 1/3 were BRCA2 mutation carriers (53 cases). In the serous and high-grade subgroups, 16.6 % and 16.8 % of cases were associated with a BRCA1 or BRCA2 germline mutation, respectively, and 17.1 % of cases harboured combined characteristics. Notably, 45 % of mutation carriers did not present a positive family history for breast and/or ovarian cancers, highlighting the fact that family history is not a sensitive marker for BRCA1/BRCA2 detection [18]. The indications for BRCA1/BRCA2 germline mutation testing in a patient with ovarian cancer are summarised in Table 2.3.
Walsh et al. used a high-throughput sequencing method to screen 21 BRCA1/BRCA2 partner genes that are candidates to be associated with an increased risk of breast or ovarian cancers in a series of 360 women with ovarian, peritoneal or fallopian tube carcinoma. Mucinous carcinomas were excluded, and a selection bias towards high-grade cases was observed, as 91 % of tumours were high grade. Among the 360 women tested, 24 % carried a deleterious mutation: 18 % in BRCA1 or BRCA2 (a figure similar to Alsop’s study) and 6 % in BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C or TP53 [19].
BRCA1, BRCA2 and HR-Associated Genes: Breast and Ovarian Cancer Risks
Two meta-analyses examined the risk of breast and ovarian cancers in BRCA1 and BRCA2 carriers [20, 21]. Note that the meta-analysis by Antoniou, performed without selection for family history, was included in the meta-analysis by Chen and Parmigiani, which combined both family and population-based studies and which mainly concerned the risks of breast and ovarian cancers (Table 2.1). Cumulative risks of ovarian cancer at ages 40, 50, 60 and 70 are reported in Table 2.2 [21]. The mean age at onset for both breast and ovarian cancers was younger in BRCA1 and BRCA2 carriers compared to the general population. In addition, according to the recent study by Alsop performed in a large series of OC women, the mean age at onset in sporadic cancer patients was 60.5 years versus 53.4 years and 59.8 years in patients with BRCA1 and BRCA2 mutations, respectively [18].
Table 2.1
70-year cumulative risk of breast and ovarian cancer in BRCA1 and BRCA2 mutation carriers
Cumulative cancer risk at age 70 | BRCA1 (95 %CI) | BRCA2 (95 %CI) |
|---|---|---|
Breast cancer | ||
Antoniou et al. (2003) | 65 % (44–78) | 45 % (31–56) |
Chen and Parmigiani (2007) | 57 % (47–66) | 49 % (40–57) |
Ovarian cancer | ||
Antoniou et al. (2003) | 39 % (18–54) | 11 % (2.4–19) |
Chen and Parmigiani (2007) | 40 % (35–46) | 18 % (13–23) |
Table 2.2
Predicted ovarian cancer risk of a 30-year-old woman carrying a BRCA1 or BRCA2 germline mutation
Risk (%) of developing ovarian cancer by age | ||||
|---|---|---|---|---|
30-year-old woman with a BRCA1/BRCA2 mutation | Risk at age 40 Mean (95 %CI) | Risk at age 50 Mean (95 %CI) | Risk at age 60 Mean (95 %CI) | Risk at age 70 Mean (95 %CI) |
BRCA1 | 2.2 (1.6–3.4) | 8.7 (6.7–12) | 22 (18–27) | 39 (34–43) |
BRCA2 | 0.52 (0.28–1) | 2.4 (1.5–4.2) | 7.4 (5.1–11) | 16 (12–20) |
These figures correspond to mean cancer risks. Shortly after the identification of BRCA1 and BRCA2, it was observed that cancer risks may differ from one family to another (defined by close relatives) and among relatives of the same family. These differences were not chance differences but were underpinned by modifying factors that can be either genetic or non-genetic or by the nature/location of the causative mutation. Two international consortia have been established in order to identify such modifying factors: HBCCS and CIMBA. A recent study performed by CIMBA in a very large number of women (19,581 BRCA1 and 11,900 BRCA2 mutation carriers) identified regions of the coding sequence in both genes in which the relative risk of ovarian cancer may be higher than the relative risk of breast cancer [22]. A genome-wide association study conducted on a series of 11,403,952 SNPs disseminated throughout the genome on 15,437 sporadic cases, 15,252 BRCA1 carriers, 8211 BRCA2 mutation carriers and 30,845 controls has also identified 6 SNPs associated with a slight increase of the relative risk of ovarian cancer. However, only two of these SNPs increase the risk of ovarian cancer in BRCA1 mutation carriers, and only one increases the risk of ovarian cancer in BRCA2 mutation carriers [23]. The results of these extensive studies are disappointing at the present time, as they do not lead to any modification of the management of at-risk women, but they need to be pursued by combining factors of various origins.
Few data are available concerning ovarian cancer risk associated with germline mutation of genes involved in HR. At the present time, estimated cancer risks are only available for two RAD51 paralogs, RAD51D and RAD51C [24, 25]. The relative risk of ovarian cancer was estimated to be 6.30 (95 % CI: 2.86–13.85) in RAD51D mutation carriers and 5.88 (95 % CI: 2.91–11.88) for RAD51C mutation carriers, which constitutes a >9 % cumulative risk by age 80 [25, 26]. The lack of precise estimates of cancer risk associated with these newly identified genes is a major limitation to their use for genetic counselling in clinical practice. However, genes involved in HR could be used to guide treatment.
Prevalence of Somatic Inactivation of BRCA1/BRCA2 and HR Genes in Ovarian Cancer and Related Diseases
Although it has been clearly demonstrated that, in the presence of a germline BRCA1/BRCA2 mutation, the second allele is somatically inactivated, identification of the BRCA1 gene immediately raised the question of its possible bi-allelic somatic inactivation. The article reporting the identification of BRCA1 in the October 1994 issue of Science was accompanied by another article reporting a study based on a series of 32 breast carcinomas selected for a deletion of the 17q arm, in which BRCA1 is located. Although three BRCA1 mutations were detected in the tumour, they all corresponded to germline mutations [27]. Consequently, up until recently, tumour inactivation of BRCA1/BRCA2 genes was considered to be mainly associated with germline mutations. However, recent studies, based on high-throughput sequencing techniques in large series of ovarian cancers, have thrown new light on this issue, which is of critical importance with the recent development of DDR inhibitors, to which strictly somatically BRCA1/BRCA2 inactivated tumours are expected to also be sensitive.
The Cancer Genome Atlas (TCGA) project selected 316 high-grade serous ovarian carcinomas. Exome, promoter methylation, transcriptome, microRNA expression and DNA copy number were studied for each tumour [28]. Germline DNA was matched. Tumour analyses identified 73 BRCA1/BRCA2 mutations (23 %), which were of germline origin in 52 cases (17 %). Conversely, in 21 (6 %) tumours, BRCA1/BRCA2 inactivation was strictly somatic. In summary, 25 % (21/73) of BRCA1/BRCA2 inactivations may be somatic. The BRCA1 promoter has also been shown to be methylated in about 10 % of tumours, suggesting loss of expression. Genes of the HR pathway (EMSY, FANC, RAD51C, PALB2, CHEK2, BRIP1) have also been found to be mutated in the absence of BRCA1/BRCA2 inactivation.
In the study by Pennington et al., providing an update to the study by Walsh et al., 30 genes, including BRCA1, BRCA2 and 13 genes involved in the HR pathway and cell cycle control (BRCA1, BRCA2, ATM, BARD1, BRIP1, CHEK1, CHEK2, FAM175A, MRE11A, NBN, PALB2, RAD51C, RAD51D), were sequenced in a series of 390 cases of high-grade OC at both germline and tumour levels [19, 29]. A deleterious BRCA1/BRCA2 mutation was identified in 24 % of tumours (18 % germline and 6 % strictly somatic). Deleterious mutations of other genes were also identified in 8.6 % of cases (6 % germline and 2.6 % strictly somatic). The somatic/germline inactivation ratio was 25 %, similar to that observed in the TCGA study (Fig. 2.2). It is noteworthy that although germline HR pathway gene mutations do exist in low-grade serous carcinoma (11 % of cases), no strictly somatic gene inactivation has been observed.
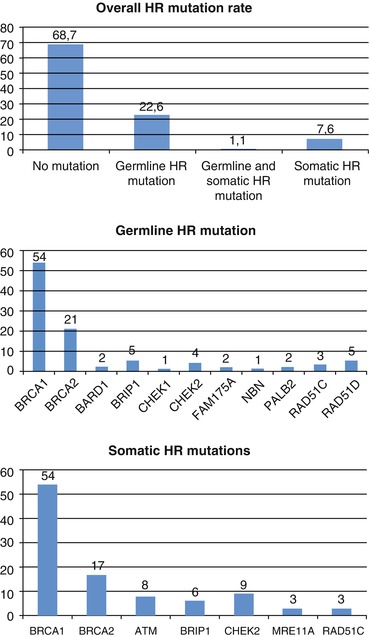
Fig. 2.2
Mutation rates in homologous recombination (HR) genes (From Pennington et al. [29]). (a) According to Pennington’s study in 367 subjects, 115 (31.3 %) had deleterious mutations in one of 13 HR genes tested: 83 (22.6 %) with germline mutations, 28 (7.6 %) with somatic mutations and 4 (1.1 %) with both germline and somatic mutations. (b) According to Pennington’s study in 367 subjects, 87 subjects (24 %) had germline mutations in 11 HR genes: 49 (13.4 %) in BRCA1, 17 (4.6 %) in BRCA2 and 22 (6 %) in other homologous recombination genes, including BARD1, BRIP1, CHEK1, CHEK2, FAM175A, NBN, PALB2, RAD51C and RAD51D. (c) According to Pennington’s study in 367 subjects, 32 carcinomas (8.7 %) had a total of 35 somatic mutations in 7 HR genes: 19 (5.2 %) in BRCA1, 6 (1.6 %) in BRCA2 and 10 (2.7 %) in other homologous recombination genes, including ATM, BRIP1, CHEK2, MRE11A and RAD51C
The Pennington study also reported that tumours demonstrating inactivation of the BRCA1/BRCA2 or HR pathway genes, regardless of its origin, are more sensitive to platinum-based therapy than non-mutated tumours [29]. Due to the complexity of genetic testing, especially on formalin-fixed, paraffin-embedded tissues, the availability of a tumour BRCAness or HRness signature would be highly desirable to select patients for clinical trials and specific treatments. Such signatures, which correspond to genomic scars of the HR defect, are currently under development [30–32] and are starting to be used in clinical trials [33].
Lynch Syndrome
Definition
Lynch syndrome, also known as hereditary non-polyposis colorectal cancer (HNPCC), was first described by Henry Lynch, who reported rare familial aggregations of colorectal, gastric, endometrium, small bowel, biliary tract, urothelium tract and ovarian cancer with early onset and whose distribution in one side of the family suggested a predisposing gene transmitted according to an autosomal dominant mode [34]. The Amsterdam clinical criteria, initially defined arbitrarily in order to select families for identifying responsible genes, should now be abandoned. The current definition of Lynch syndrome is molecular, based on identification of an inactivating monoallelic germline mutation in a gene involved in the DNA mismatch repair pathway (MMR): MLH1, MSH2, MSH6 or exceptionally PMS2 [35]. As indicated for BRCA1 and BRCA2, Lynch syndrome is associated with a marked heterogeneity of deleterious mutations. In addition, there are also a large number of variants of unknown significance that require complementary classification studies.
Function of the Mismatch Repair Pathway
The function of the MMR pathway is to correct DNA polymerase nucleotide misincorporations that may occur during DNA replication. Seven proteins compose the human MMR system with three MutS-homologs (MSH2, MSH3 and MSH6) and four MutL homologs (MLH1, MLH3, PMS1 and PMS2). MutS proteins recognise a mismatch and recruit the ATP-bound MutL protein and then correct the mismatch. The MutS homodimer is formed by either MSH2/MSH6 (the MutSα complex) for single-base mismatches and short insertion–deletion loops or MSH2/MSH3 (the MutSβ complex) for larger loops. The endonuclease function in the PMS2 subunit of MutLα (formed by MLH1 and PMS2) excises the DNA strand containing the wrong nucleotide and resynthesises the excision gap via the replicative DNA polymerase.
In MMR pathway-deficient cells, short tandem repeat sequences, i.e. microsatellites, appear particularly prone to nucleotide misincorporations. The resulting microsatellite instability (MSI) is a hallmark for MMR defects (for review, see [36]). Lynch syndrome with genome-wide microsatellite instability therefore presents a signature of MMR dysfunction. This signature is applied in routine diagnosis. This signature is sensitive – the absence of MSI can almost formally exclude the diagnosis of Lynch syndrome (sensitivity of about 90 %, but less reliable with MSH6) – and nonspecific, as MSI may result from MLH1 promoter methylation in late-onset colorectal cancers [37, 38]. Techniques and interpretation are now well standardised. Immunohistochemistry (IHC) analyses of MMR protein expression should also be performed, as the mutated gene is expected to lead to loss of expression of the corresponding protein in tumour tissue, which may guide genetic screening [38].
Related posts:
 Treatment at Relapse (Surgery and Systemic Treatments)
Serous Tumors of Low Malignant Potential and Low-Grade Serous Carcinomas of the Ovary or Peritoneum
High-Grade Carcinomas, BRCA Mutations and the Role of PARP Inhibitors
Mucinous Carcinoma of the Ovary
Treatment at Relapse (Surgery and Systemic Treatments)
Serous Tumors of Low Malignant Potential and Low-Grade Serous Carcinomas of the Ovary or Peritoneum
High-Grade Carcinomas, BRCA Mutations and the Role of PARP Inhibitors
Mucinous Carcinoma of the Ovary
 Ovarian Sex Cord Tumors
Ovarian Sex Cord Tumors
 Malignant Ovarian Germ Cell Tumours: An Overview of Management and Controversies
Malignant Ovarian Germ Cell Tumours: An Overview of Management and Controversies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


