Distribution of gliomas based on histologic subtype in the USA. Glioblastoma multiforme accounts for the majority of gliomas (54.5%). Astrocytomas of all grades, including glioblastoma multiforme, account for approximately 75% of all gliomas. NOS, not otherwise specified. (Based on statistics from 2007 to 2011 collected by Central Brain Tumor Registry of the United States [CBTRUS].)
Grade I pilocytic astrocytoma
Pilocytic astrocytoma is primarily a pediatric disease, but it also affects young adults. This tumor commonly arises in the cerebellum and is characterized as a slow-growing, well-circumscribed mass that rarely progresses to a higher-grade lesion and responds well to surgical resection.2 Pilocytic astrocytomas are unique among gliomas as they usually possess few genetic alterations. These alterations often lead to activation of a single pathway, the mitogen-activated protein kinase (MAPK) pathway. The most common alteration is KIAA1549-BRAF (73%), a chromosomal tandem duplication that results in the formation of a constitutively active fusion protein. Other alterations found in pilocytic astrocytoma include BRAF point mutations (V600E) and additional BRAF fusions that result in constitutive kinase activation. Inactivating mutations in the gene NF1 are also seen in pilocytic astrocytoma, usually in patients affected by neurofibromatosis type 1 (NF1), who have existing germline mutations and are at increased risk for developing these tumors.6
Grade II–III ependymoma
Ependymomas arise from ependymal cells that line the ventricular system and spinal cord, and account for 7% of all gliomas.1 In children, these tumors most often arise intracranially (90%), while in adults they tend to localize to the spinal cord. Spinal cord ependymomas have a better outcome than intracranial lesions; however, the delicate location of the tumor can make surgical resection challenging and leave patients with neurological deficits.7 Ependymomas are grade II tumors that are slow-growing and generally amenable to surgical excision, while grade III anaplastic ependymomas are proliferative tumors that tend to infiltrate the surrounding tissues or metastasize along the ventricular system.2 Amplification of chromosome 7 and loss of chromosome arm 22q are the most common genetic alterations in adult ependymoma.2 Fifty percent of adults with spinal cord ependymoma have mutations in the neurofibromatosis 2 (NF2) gene, located on 22q, and an increased incidence of ependymoma is seen in patients with the disease neurofibromatosis type 2, who have germline mutations in NF2.2,7
Grade II and III astrocytoma: diffuse and anaplastic astrocytoma
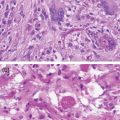
Diffuse and anaplastic astrocytomas together account for 12% of all primary CNS malignancies.1 These tumors commonly localize to the cerebral hemispheres and are characterized by indistinct borders with microscopic extensions that spread diffusely into the surrounding tissue. The invasive nature of these tumors makes them difficult to resect completely, predisposing patients to tumor recurrence and progression. Histologically, grade II diffuse astrocytomas are composed of well-differentiated astroglial tumor cells with mildly atypical nuclei and increased cellularity. Grade III anaplastic astrocytomas are characterized by the presence of mitotic figures, as well as increased nuclear atypia and cellularity relative to diffuse astrocytomas.2 Grade II and III astrocytomas can present de novo and both tend to progress to higher grades (II→III or II/III→IV). However, not all patients survive to the point of developing a grade IV secondary GBM, reflected in part by the incidence rate of grade II and III astrocytomas being two to three times greater than that of secondary GBM.8 The progression from low-grade (II) astrocytoma to secondary GBM takes an average of 5 years; therefore, efforts are made to treat and closely monitor lower-grade gliomas for progression (Figure 1.2).2
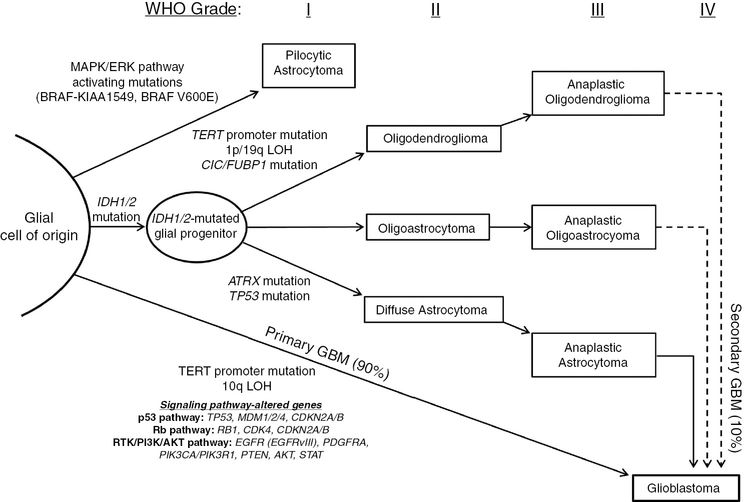
Common genetic alterations in glioma tumorigenesis and progression. Grade I gliomas, known as pilocytic astrocytomas, are almost all characterized by MAPK pathway activation, frequently due to BRAF-KIAA1549 fusion transcripts. At the opposite end of the spectrum are primary GBMs, found to have characteristic TERT promoter mutations and alterations in key signaling pathways such as the p53, Rb and RTK/PI3K/AKT pathways. Most progressive gliomas have mutations in IDH1/2, which occur early in gliomagenesis, with the potential to develop into secondary GBM. Though it is unclear if the cell of origin is the same for all glioma subtypes, IDH1/2 mutations occur early in gliomagenesis, and secondary alterations likely dictate development into oligodendroglioma (TERT promoter mutations, 1p/19q loss of heterozygosity, CIC/FUBP1 mutation), diffuse astrocytoma (ATRX mutation, TP53 mutation), or mixed oligoastrocytoma. Each of these can progress to anaplastic lesions (grade III), and eventually result in secondary GBM (grade IV).
Compared to diffuse astrocytomas, anaplastic lesions present in older patients (median age of diagnosis 53 vs. 48 years) and have a poorer prognosis (5-year survival 27.3% vs. 47.4%) (Figure 1.3).9 Both grade II and III astrocytomas are characterized by a particular genetic signature, consisting of frequent (>75%) co-occurring mutations in the genes IDH1/2, ATRX, and TP53 (Figure 1.4).
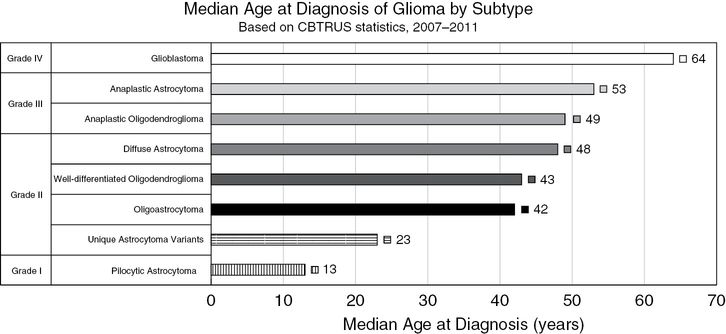
Median age of glioma patients in the USA based on histologic subtype. A general trend is observed across glioma subtypes in which higher-grade gliomas are more commonly diagnosed in older populations. Ependymomas have been excluded because grade-specific statistics were unavailable; however, the median age at diagnosis of grade I–III ependymomas collectively is 44 years. (Based on statistics from 2007 to 2011 collected by Central Brain Tumor Registry of the United States [CBTRUS].)

Common genetic alterations in glioma occur in distinct patterns unique to each histology, defining molecular subtypes of glioma. Primary GBM is largely characterized by TERT promoter mutations, while secondary GBM has a mutational spectrum similar to those seen in lower-grade astrocytomas, including co-occurring mutations in IDH1/2, ATRX, and TP53. Oligodendrogliomas have co-occurring mutations in IDH1/2, the TERT promoter, and concurrent loss of 1p19q. While oligoastrocytomas have mixed histologic patterns, genetically they resemble the patterns of either astrocytoma or oligodendroglioma.
Grade II and III oligodendroglioma: well-differentiated and anaplastic oligodendroglioma
Oligodendrogliomas account for 4.9% of all primary malignant brain tumors. The population affected by oligodendroglioma is generally younger than that of astrocytoma, with the median age of diagnosis for grade II and III oligodendroglioma being 43 and 49 years, respectively (Figure 1.3).1 In addition, oligodendrogliomas are generally less aggressive tumors than astrocytomas, with 79.5% of grade II and 52.2% of grade III oligodendroglioma patients surviving past 5 years.9 Oligodendrogliomas frequently arise in the frontal lobes of the cerebral hemispheres and form microcalcifications around neurons and blood vessels that can occasionally can be seen on radiographic imaging. Histologically, oligodendrogliomas are characterized by hypercellular masses of uniform and infiltrating cells with rounded nuclei and perinuclear halos, an artifact of formalin fixation and paraffin embedding that gives a characteristic “fried egg” appearance. Well-differentiated oligodendrogliomas (grade II) can evolve into high-grade (grade III) anaplastic oligodendrogliomas, which have increased mitotic activity, vascular proliferation, and necrosis.2 Oligodendrogliomas most frequently harbor co-occurring IDH1/2 mutations, TERT promoter mutations, 1p/19q chromosomal loss, and alterations in the genes encoding the transcriptional regulators CIC and FUBP1 (Figure 1.4).4,10,11
Grade II and III oligoastrocytoma: oligoastrocytoma and anaplastic oligoastrocytoma
Oligoastrocytomas have histologic features of both astrocytomas and oligodendrogliomas: either diffusely intermixed or, less commonly, as biphasic tumors with these elements separated into distinct areas. Historically, the diagnosis of oligoastrocytoma has posed a challenge for neuropathologists due to poorly defined criteria for classification and subjectivity in interpretation of histologic features.2 Although collectively oligoastrocytoma patients have a 5-year survival of 61.1%, there is substantial clinical variability.2 Recent work has helped ameliorate this histologic and clinical ambiguity by revealing a genetic delineation of oligoastrocytomas into two distinct groups: oligoastrocytomas with co-occurring mutations in ATRX, IDH1/2, and TP53, and oligoastrocytomas with co-occurring mutations in the TERT promoter, IDH1/2, and 1p/19q loss of heterozygosity, reflecting the genetic patterns of astrocytomas and oligodendrogliomas, respectively (Figure 1.4).4 These genetic associations are evident clinically, as oligoastrocytoma patients with 1p/19q co-deletion have a longer progression-free survival than those with intact 1p/19q (60 vs. 30 months), paralleling the generally better prognosis of patients with oligodendrogliomas when compared to those with astrocytomas.12
Grade IV glioblastoma: primary and secondary glioblastoma
GBM is the most common malignant brain tumor, representing almost half (45.6%) of all primary CNS malignancies. GBM is also one of the deadliest cancers, with an average survival of 12 months after diagnosis and a 5-year-survival of <5.0%.1,4 Histologically, they are characterized by highly anaplastic cells with frequent mitoses, vascular proliferation, and necrosis. As mentioned previously, GBMs can develop in two different ways: either de novo as primary GBMs without evidence of a lower-grade precursor glioma, or as secondary GBMs, which develop from lower-grade gliomas (II–III). While histologically these two types of GBM are largely indistinguishable, there are key differences between primary and secondary GBM in terms of the populations they affect, their clinical behavior, and the underlying genetic alterations that drive their development. Primary GBM, accounting for >90% of GBMs, typically arises in older patients with a mean age of 62 years, while secondary GBM, which accounts for the remaining 5–10% of cases, has a mean age at diagnosis of 45 years.2 While it usually takes several years for secondary GBM to develop from a lower-grade astrocytoma, most patients with primary GBM experience their first symptoms only 3 months before diagnosis.13 Secondary GBM is also inherently less aggressive, with patients exhibiting a better overall survival from the time of diagnosis than patients with primary GBM.2
Until recently, clinical history was the primary method of differentiating between primary and secondary GBM, due to their histologic similarity. However, recent genomic analyses have revealed that these two subtypes of GBM are not only clinically, but also genetically, distinct diseases. Primary GBM is most clearly distinguished by TERT promoter mutations in the absence of IDH1/2 mutations, while secondary GBM is characterized by co-occurring mutations in IDH1/2, ATRX, and TP53, without TERT promoter mutations (Figure 1.4).4,11 Other alterations found in primary GBM, such as EGFR amplification, 10q loss of heterozygosity (LOH), and CDKN2A deletion, will be discussed in the following sections.
Common genetic alterations in glioma
There have been a number of studies aimed at understanding the alterations that drive gliomagenesis from a genetic, epigenetic, and transcriptional viewpoint. While many of these investigations have focused on GBM, the findings have also provided insight into lower-grade gliomas, highlighting the importance of these alterations to glioma initiation and progression. This section outlines many of the significant discoveries resulting from these efforts, starting with the major signaling pathway alterations and transcriptional subtypes of GBM, followed by an in-depth discussion of the recurrent genetic mutations and their functions in glioma, including alterations in IDH1/2, the MGMT promoter, 1p/19q, CIC/FUBP1, ATRX, and the TERT promoter.
Signaling pathway alterations in glioblastoma
In 2008, groups at Johns Hopkins University, Duke University, and The Cancer Genome Atlas (TCGA), carried out large-scale investigations using genetic sequencing, copy number analysis, and gene expression profiling to identify the major genetic alterations in GBM.3,14 Three dominant signaling pathways were found to be affected by somatic mutation, gene amplification, and/or homozygous deletion: the p53 (TP53, MDM2, MDM4), Rb (RB1, CDK4/6 and CDKN2A/B), and RTK/PI3K/PTEN (EGFR, PDGFRA, PIK3CA, PIK3R1, PTEN, AKT) pathways. A subsequent study by the TCGA in 2013 confirmed and expanded on many of these findings.15 It is worth noting that these pathways have substantial overlap, such as CDKN2A (Rb pathway) and AKT (RTK/PI3K/PTEN pathway) both affecting MDM2 levels (p53 pathway), illustrating that single alterations can have complex and far-reaching effects on multiple cellular processes.
Genetic alterations in the p53 pathway: TP53 and CDKN2A/MDM2/MDM4
Alterations to the p53 pathway are found in the vast majority of GBMs (86%), with deletion or mutation of TP53 (27.9%), amplification of MDM1/2/4 (15.1%), and homozygous deletion of CDKN2A/B (61%) occurring in a largely mutually exclusive fashion.15 p53 is a tumor suppressor encoded by the TP53 gene and that functions as a critical regulator of the mammalian cell cycle, promoting repair pathway activation, growth arrest, or apoptosis in response to stressors such as DNA damage, oxidative stress, and oncogene activation. Though tumor suppressors generally require loss of both alleles in order to lose regulatory control, TP53 mutations can act in a dominant negative fashion, wherein a heterozygous mutation is sufficient to compromise native p53 regulatory function and promote oncogenesis. MDM2/4 and p53 interact through an auto-regulatory negative-feedback loop in which p53 activates transcription of MDM2/4, which in turn targets p53 for ubiquitin-mediated proteasomal degradation, limiting p53’s growth-suppressive activity in the absence of cellular stress. CDKN2A encodes two proteins through alternate reading frames p16Ink4A and p14ARF. p14ARF responds to over-activation of growth signaling pathways by inhibiting MDM2, allowing p53 to accumulate within the cell, inducing cell cycle arrest and/or apoptosis.
Genetic alterations in the Rb pathway: RB1, CDK4, and CDKN2A/B
The Rb pathway is altered in 79% of GBMs, with inactivating alterations in RB1 (7.6%), amplifications of CDK4 (14%), and homozygous deletions of CDKN2A/B (61%) occurring with high mutual exclusivity.15 RB1 loss of heterozygosity is also found in approximately 40% of GBMs. Rb is a tumor suppressor that normally binds and inactivates E2F transcription factors, which are potent drivers of G1→S cell cycle progression. Rb is phosphorylated by cyclin-dependent kinases, CDK4 and CDK6, blocking the ability of Rb to bind E2F and allowing cell cycle progression. In addition to its role in the p53 pathway described above, CDKN2A is also involved in the Rb pathway, where P16INK4A inhibits cyclin-dependent kinases, leaving Rb bound to E2F and halting the cell cycle. The high frequency of CDKN2A deletions and its regulatory role in both the p53 and Rb pathways indicate the importance of CDKN2A loss of function as a mechanism for GBM tumorigenesis.
Genetic alterations in the RTK/PI3K/AKT pathway: EGFR, PDGFRA, PIK3CA, PIK3R1, PTEN, AKT
Alterations to the RTK/PI3K/AKT pathway occur in 90% of GBMs, with 39% of cases bearing two or more alterations in this pathway. Amplification of genes that encode receptor tyrosine kinases (RTKs) occurs in 66% of GBMs (EGFR 57.4%; PDGFRA 13.1%; FGFR 3.2%; MET 1.6%), leading to increased activation of the PI3K/AKT signaling pathway.15 Normally, cells depend on RTKs to respond to external growth factor cues to activate pathways that stimulate cell growth and proliferation. However, cancer cell proliferation is innately uncoupled from such external cues, allowing tumors to grow in the absence of environmental proliferative signals. In GBM, this independence is accomplished through alterations of RTKs, such as gene amplification and/or mutations that lead to receptor auto-activation. EGFR amplification is the most common of these, occurring in more than half of GBMs. Among EGFR-amplified tumors, many also have EGFR mutations, the most common of which is an in-frame deletion of exons 2–7 that results in a truncated, constitutively active receptor variant known as EGFRvIII. GBM with EGFRvIII (and EGFR overexpression) are among the most aggressive GBMs and show prominent activation of STAT signaling. Recent work indicates that EGFR co-localizes with and phosphorylates EGFRvIII, which in turn phosphorylates the oncogenic transcription factor STAT3, suggesting a possible role for the co-occurrence of EGFRvIII and EGFR amplification in GBM.16
Alterations affecting the downstream targets of RTK signaling include mutation or deletion of PTEN (41%) and mutation or amplification of PIK3CA/PIK3R1 (25%). Normally, activation of RTKs increases PI3K activity, which phosphorylates the phospholipid PIP2 to generate PIP3, a powerful membrane-bound, intracellular signaling molecule that activates several fundamental cellular processes via AKT activation. The phosphatase and tensin homologue (PTEN) protein directly counteracts the actions of PI3K by dephosphorylating PIP3. Heterozygous loss of chromosome 10q is seen in most GBMs (>90%), and, as PTEN is located in this region, it is one of the most frequently targeted tumor suppressors in GBM.15
The transcriptional subtypes of glioblastoma
Several large-scale studies have provided evidence that GBM can be classified according to gene expression patterns.15,17,18 These classifications have been refined over time, with the current system incorporating four groups: classical, proneural, neural, and mesenchymal.17 The classical group exhibits the highest frequency of EGFR alterations, including EGFR amplification, EGFRvIII and other mutations in the receptor. In addition, they are characterized by frequent CDKN2A/B deletions, a lack of TP53 mutations, high expression levels of notch and sonic hedgehog pathway components and broad amplifications of chromosomes 19 and 20. The proneural group exhibits high expression levels of the oligodendroglial development genes PDGFRA, OLIG2, and NKX2-2, and consists of two primary subgroups based on DNA methylation patterns: G-CIMP and non-G-CIMP (glioma-CpG island methylator phenotype, detailed in epigenetics section, below). The G-CIMP group of proneural GBMs has mutations in IDH1, TP53, ATRX, and MYC, while the non-G-CIMP subtype has TP53 mutations and amplifications in PDGFRA, CDK4, SOX2, and EGFR. The neural group, consisting of the oldest patients on average, has no unique genetic alterations compared to the other subtypes, but is characterized by expression of genes typically seen in neurons, such as NEFL, GABRA1, SLC12A5, and SYT1. The mesenchymal GBMs display an expression pattern typical of mesenchymal cells, particularly markers CHI3L1 and MET. This group is observed to have frequent mutations in NF1, PTEN, and TP53.15,17
The expression patterns observed in these subtypes provide insights into the connection between common genetic alterations in GBM and their effects on the transcriptome. The following sections provide an in-depth description of the most common mutations in glioma and outline a newer, simplified molecular classification system that stratifies patients into clinically relevant groups.
IDH1 and IDH2 mutations in progressive glioma
Perhaps the most significant result of the 2008 genomic sequencing study, carried out by scientists at Duke University and Johns Hopkins University, was the discovery of recurrent missense mutations in a single codon of the gene IDH1.14 The alteration found in IDH1 was highly frequent in secondary GBM, and was associated with younger age and co-occurring mutations in TP53. IDH1 and its isoform IDH2 are involved in cellular energy production and catalyze identical reactions, however, IDH1 functions in the cytoplasm and peroxisomes, while IDH2 is localized to the mitochondria. Because secondary GBMs progress from lower-grade tumors, the next critical step was to perform a large-scale investigation of IDH1 and IDH2 in lower-grade gliomas. After analyzing over 400 CNS tumors, mutations in IDH1/2 were identified in over 80% of grade II and III astrocytomas, oligodendrogliomas, and secondary GBMs, establishing it as a highly frequent mutation, and a critical diagnostic marker of diffuse gliomas.5
The discovery of these mutations has clear clinical relevance, as patients with IDH1/2-mutant tumors have significantly longer median overall survival than those with IDH1/2-wildtype tumors in the case of both GBM (31 vs. 15 months) and anaplastic astrocytoma (65 vs. 20 months). Furthermore, IDH1/2 mutations provide a clear marker for discriminating between subtypes of GBM, as mutations are rare in primary GBM (<5%) but very common in secondary GBM (85%) (Figure 1.4).5 Additional investigations were subsequently conducted to assess the mutation status of IDH1/2 in other neoplasms. Frequent IDH1/2 mutations were discovered in acute myeloid leukemia (AML), cartilaginous tumors (i.e., Ollier disease, Maffucci syndrome), and intrahepatic cholangiocarcinoma.19–21
In glioma, mutations in IDH1 affect the arginine residue, R132, with the most frequent alteration (91%) being a single-basepair substitution of adenine for guanine, changing the encoded amino acid arginine to histidine (R132H), although there are additional rare mutations, including R132C, R132S, R132G, R132L, R132V, R132P (collectively <8%).22 Mutations in glioma are also found in IDH2 at residue R172, the analogous locus of R132 in IDH1. Although rare (2% of all IDH1/2 mutations in glioma), IDH2 R172 mutations produce similar phenotypic results and are mutually exclusive with IDH1 R132 mutations.5 IDH2 R172K (arginine for lysine) is the most common IDH2 alteration in glioma; however, other less frequent mutations have been identified at this locus, including R172M, R172W, and R172G. Interestingly, AML exhibits a different IDH1/2 mutation pattern than glioma, with mutations occurring more frequently in IDH2 than IDH1, and at residue R140, also in the enzyme’s active site, more often than at R172.23
IDH1/2 mutations cause the accumulation of the oncometabolite D-2HG in glioma
IDH1 and IDH2 are enzymes that catalyze the reversible oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG, also known as 2-oxoglutarate) while producing CO2 and reducing the cofactor NADP+ to NADPH. The commonly mutated residues, R132 of IDH1 and R172 of IDH2, are located in the enzymatic active site and form hydrogen bonds with the substrate isocitrate. Although IDH1/2 mutations were initially thought to confer a loss of function, subsequent work demonstrated that the IDH1 mutant enzyme exhibits decreased affinity for isocitrate and NADP+ and increased affinity for α-KG and the cofactor NADPH.5,24 Most importantly, it was discovered that this change in substrate specificity gives mutant IDH1 the neomorphic ability to catalyze the NADPH-dependent reduction of α-KG, forming the oncometabolite D-2-hydroxyglutarate (D-2HG), in a stereospecific manner (Figure 1.5). This discovery was confirmed in human glioma samples by mass spectrometry, where tissues with IDH1 R132 mutations had 100-fold excess levels of D-2HG compared to IDH1-wildtype tumors.25 Further studies extended these findings to the analogous IDH2 mutations, revealing elevated levels of D-2HG in samples from AML patients bearing IDH2-active site mutations (R172 and R140).23
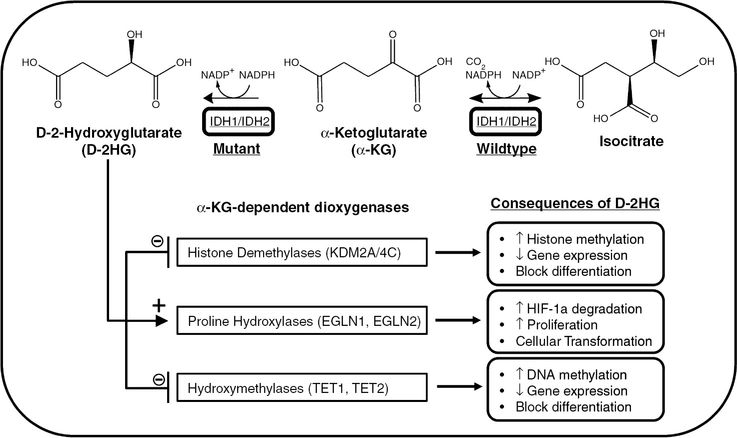
IDH1/2 mutations confer neomorphic enzymatic activity, resulting in the production of D-2-hydroxyglutarate (D-2HG). Accumulation of D-2HG, which is structurally similar to α-ketoglutarate (α-KG), interferes with several α-KG-dependent processes within the cell. D-2HG-mediated inhibition of hydroxymethylases and histone demethylases leads to the respective hypermethylation of DNA and histones, resulting in decreased transcription of genes required for differentiation. D-2HG also acts in an enantiomer-specific manner to increase proline hydroxylase activity and degradation of hypoxia-inducible factor-1α (HIF-1α), associated with cell proliferation and transformation.
Both IDH1 and IDH2 function natively as homodimers, and as IDH1/2 mutations are almost always heterozygous, it was speculated the mutant IDH1/2 enzymes could bind their wildtype counterparts, forming heterodimers. Prior to the discovery of the neomorphic activity of mutant IDH1/2, studies found decreased NADPH levels in IDH1-mutant cells, leading to the hypothesis that a dominant-negative interaction between the mutant and wildtype enzymes was occurring.24 It was later shown, however, that mutant/wildtype heterodimer formation was specific to IDH1, and not IDH2, diminishing the likelihood that this phenomenon was the primary mechanism for the oncogenic effect of mutant IDH1/2.26 Subsequent work has shown that mutant IDH1 profits from close association with wildtype IDH1 as a source of α-KG and NADPH, which it quickly utilizes for production of D-2HG.27 Accordingly, loss of wildtype IDH1 results in lower levels of D-2HG produced within IDH1-mutant tumors.28 In the case of mutant IDH2, loss of the wildtype allele does not have the same effects likely due to the abundance of α-KG in the mitochondria.29 Importantly, it was demonstrated that the oncogenic effects of mutant IDH1/2 can be fully recapitulated in IDH1/2-wildtype cells by exogenous administration of cell-permeable D-2HG, further confirming the role of D-2HG as an oncometabolite.30
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


