The Biological Nature of Intrinsic Aging
Understanding the biology of intrinsic aging is important for geriatricians because of the insights it provides into: (i) why and how the body becomes progressively more vulnerable to disability and disease as we grow older; (ii) how we might intervene in the underlying mechanisms; and (iii) what is the exact nature of the relationship between “normal” aging and age-related diseases. Although there is significant variability in how aging affects individuals, certain underlying processes appear to follow a common course. Furthermore, humans share some features of aging with a wide range of other animal species. Thus, even though much of the research on intrinsic aging is done using simple animal models such as the roundworm Caenorhabditis elegans or the fruitfly Drosophila melanogaster, it appears that common regulatory pathways are at least partially conserved across the spectrum that includes mammals, and in particular humans. This is especially the case for fundamental mechanisms that protect cells against shared threats such as damage to DNA arising from endogenous stressors like reactive oxygen species (ROS), which are essential molecular by-products of the body’s dependence on oxygen to provide energy.
One of the central questions in the biology of aging is the nature of the genetic contribution to longevity. How do genes act on the aging process, and how does an individual’s genetic endowment contribute to their longevity?
Aging and longevity are clearly influenced by genes. Firstly, the life spans of human monozygotic twin pairs are more similar than life spans of dizygotic twins. Secondly, there are differences in life span between different genetically inbred strains of any given laboratory animal, such as the mouse. Thirdly, studies of simple organisms like fruit flies, nematode worms, and yeast have identified gene mutations that affect duration of life. However, although genes influence longevity, genes appear to account for only about 25% of the variance in human life span.
The nature of the genetic contribution to the aging process has received much attention, both from the perspective of evolutionary theory and through experimentation. The evolutionary angle is valuable because it can tell us a great deal about the kinds of genes that are likely to underlie the aging process. A commonly held belief is that aging evolved as an evolutionary necessity—for instance, to remove older individuals who might otherwise consume resources needed by the young. However, there is little support for the idea that aging does actually fulfill such a role. In nature, the vast majority of animals die young, long before they could become obstructive to the interests of the next generation. Out of a population of newborn wild mice, for example, nine out of 10 of them will be dead before 10 months even though half of the same animals reared in captivity would still be alive at 2 years. Thus, aging in mice is seen only in protected environments, and until relatively recently the same would have been true for human populations. It is only in the last 200 years that human life expectancy, even in the most advantaged countries, exceeded 40 years.
Since it is rare that an animal survives long enough to show any sign of aging, a genetically programmed means to limit population size and avoid overcrowding, if it exists, is not often brought into play. Furthermore, the idea that aging could be beneficial needs to be set against the fact that even if an animal did reach older age, the adverse effects of intrinsic senescence on fertility and risk of dying are far from beneficial as far as the individual is concerned. The idea of programmed aging rests on the notion that organisms should weaken and die altruistically, for the “good of the species.” While there may be instances where natural selection has produced apparently altruistic outcomes, evolutionary biologists have carefully delineated the rather special circumstances required for this kind of selection force to work. Aging does not fit these requirements. The argument against programmed aging can be summarized by observing that if such a program did exist, what would prevent a mutation occurring that abrogated the program and allowed the mutant to survive indefinitely? The fact that such mutants are never observed is empirical support that backs up the other objections.
The evidence from nature is that instead of being programmed to die, organisms are genetically programmed to survive. However, in spite of a formidable array of survival mechanisms, most species appear not to be programmed well enough to last indefinitely. A key to understanding why this should be so, and what governs how long a survival period there should be, comes from studying survival patterns in wild populations. If 90% of wild mice are dead by the 10 months, any investment in programming for survival much beyond this point can benefit at most 10% of the population. This immediately suggests that there will be little evolutionary advantage in programming long-term survival capacity into a mouse. The argument is further strengthened by the observation that nearly all of the survival mechanisms required by the mouse to combat intrinsic deterioration (DNA damage, protein oxidation, etc.) require metabolic resources. Metabolic resources are scarce, as is evidenced by the fact that the major cause of mortality for wild mice is cold, due to insufficient energy to maintain body temperature. From a Darwinian point of view, the mouse will benefit more from investing any spare resource into thermogenesis or reproduction than into better DNA repair capacity than its need for longevity.
This concept, with its explicit focus on evolution of optimal levels of cell maintenance, is termed the “disposable soma” theory. In essence, the investments in durability and maintenance of somatic (nonreproductive) tissues are predicted to be sufficient to keep the body in good repair through the normal expectation of life in the wild environment, with some measure of reserve capacity. Thus, it makes sense that mice (with 90% mortality by 10 months) have intrinsic life spans of around 3 years, while humans (who probably experienced something like 90% mortality by the age of 50 years in our ancestral environment) have intrinsic life spans limited to about 100 years. The distinction between somatic and reproductive tissues is important because the reproductive cell lineage, or germ line, must be maintained at a level that preserves viability across the generations, whereas the soma needs only to support the survival of a single generation.
This concept helps to explain why species differ in their intrinsic life spans. It is noteworthy, for example, that species of birds and bats tend to have greater longevity than flightless species. The reason is thought to be that flight is a very successful way of reducing an animal’s mortality risk, since it is possible to escape many predators and to forage over a greater range, thereby lessening the risk of starvation caused by a local food shortage. If risk is reduced, the statistical chance of living a bit longer is increased, and so it becomes worthwhile to invest in a higher level of somatic maintenance and repair. Studies comparing the biochemistry of cellular repair among long- and short-lived species bear this prediction out. Cells from long-lived organisms exhibit greater capacity to repair molecular damage and withstand biochemical stresses than cells from short-lived species.
The disposable soma theory provides a bridge between understanding not only why aging occurs but also how aging is caused in molecular and cellular terms. It thus extends earlier considerations by Medawar, who suggested that because organisms die young there is little force of selection to oppose the accumulation within the genome of mutations with late-acting deleterious effects, and by Williams, who suggested that genes with beneficial effects would be favored by selection even if these genes had adverse effects at later ages. This is known as the theory of “antagonistic pleiotropy,” the term pleiotropy meaning that the same gene can have different effects in different circumstances.
The current synthesis of evolutionary ideas about why aging occurs can thus be summarized in four points:
There are no specific genes for aging.
Genes of particular importance for aging and longevity are those governing durability and maintenance of the soma.
There may exist other genetically determined trade-offs between benefits to young organisms and their viability at older ages.
There may exist a variety of gene mutations with late deleterious effects that contribute to the senescent phenotype.
Given these points, it is clear that multiple genes contribute to the aging phenotype. A cornerstone of the current research agenda is to identify these genes and discover which are the most important.
Aging appears to be driven by the progressive accumulation through life of a variety of random molecular defects that build up within cells and tissues. These defects start to arise very early in life, probably in utero, but in the early years, both the fraction of affected cells and the average burden of damage per affected cell are low. However, over time the faults increase, resulting eventually in age-related functional impairment of tissues and organs (Figure 2-1). This concept makes clear the life-course nature of the underlying mechanisms. Aging is a continuous process, starting early and developing gradually, instead of being a distinct phase that begins in middle to late life.
Figure 2-1.
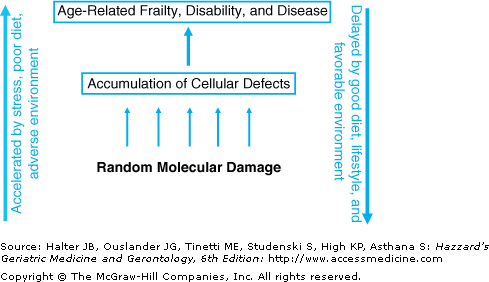
The aging process is driven by a lifelong accumulation of molecular damage, resulting in gradual increase in the fraction of cells carrying defects. After sufficient time has passed, the increasing levels of these defects interfere with both the performance and functional reserves of tissues and organs, resulting in age-related frailty, disability, and disease. Stress, adverse environment, and poor nutrition can increase the rate at which molecular damage arises. Intrinsic maintenance mechanisms, such as DNA repair and antioxidants, can slow the rate of accumulation.
Since there are multiple kinds of molecular and cellular damage, and a corresponding variety of mechanisms to protect against and repair them, aging is a highly complex process involving multiple mechanisms at different levels. This multiplicity of candidate mechanisms can easily become confusing, particularly since those researching a specific mechanism have a tendency to suggest that their preferred choice is the mechanism that drives aging. It will be apparent from the previous section that, in fact, it is highly unlikely that any single mechanism can tell the whole story. We will consider the prominent hypothesized mechanisms below. For each mechanism, there is evidence supporting the hypothesis that it is indeed an agent of senescence. However, the extent of the contribution to senescence is generally too small for the mechanism to be a sufficient explanation of age-related degeneration. The clear inference is that aging is multicausal and that the various mechanisms all play their part and are likely to interact synergistically. For example, a build-up of mitochondrial DNA mutations will lead to a decline in the cell’s energy production, and this will reduce the capacity to carry out energy-dependent protein clearance. In recent years, novel methods based on computer modeling of interactions and synergism between different aging mechanisms have begun to build a better integrated picture of how cells break down with age and there is growing experimental evidence that interactions between multiple mechanisms are important.
An important theme linking several different kinds of damage is the action of ROS (also known as free radicals), which are produced as by-products of the body’s essential use of oxygen to produce cellular energy. Of particular significance are the contributions of ROS-induced damage to cellular DNA through (i) damage to the chromosomal DNA of the cell nucleus, resulting in impaired gene function, (ii) damage to telomeres—the protective DNA structures that appear to “cap” the ends of chromosomes (analogous to the plastic tips of shoelaces), and (iii) damage to the DNA that exists within the cell’s energy-generating organelles, the mitochondria, resulting in impaired energy production.
Damage to DNA is particularly likely to play a role in the lifelong accumulation of molecular damage within cells, since damage to DNA can readily result in permanent alteration of the cell’s DNA sequence. Cells are subject to mutation all the time, both through errors that may become fixed when cells divide and as a result of ROS-induced damage, which can occur at any time. Numerous studies have reported age-related increases in somatic mutation and other forms of DNA damage, suggesting that an important determinant of the rate of aging at the cell and molecular level is the capacity for DNA repair.
A key player in the immediate cellular response to DNA damage is the enzyme poly(ADP-ribose) polymerase-1 (PARP-1). There is a strong, positive correlation of PARP-1 activity with the life span of the species: cells from long-lived species having higher levels of PARP-1 activity than cells from short-lived species. Similarly, human centenarians, who have often maintained remarkably good general health, appear to have a greater poly(ADP-ribosyl)ation capacity than the general population.
In many human somatic tissues, a decline in cellular division capacity with age appears to be linked to the fact that the telomeres, which protect the ends of chromosomes, get progressively shorter as cells divide. This is due to the absence of the enzyme telomerase, which in humans is normally expressed only in germ cells (in testis and ovary) and in certain adult stem cells. Some studies have suggested that in dividing somatic cells, telomeres act as an intrinsic “division counter,” perhaps to protect us against runaway cell division as happens in cancer but causing aging as the price for this protection. Erosion of telomere length below a critical length appears to trigger activation of cell cycle “checkpoints,” especially the p53/p21/pRb system, resulting in permanent arrest of the cell’s capacity for further division.
The loss of telomeric DNA is often attributed mainly to the so-called “end-replication” problem—the inability of the normal DNA copying machinery to copyright to the very end of the strand in the absence of telomerase. However, biochemical stress has an even bigger effect on the rate of telomere loss. Telomere shortening is greatly accelerated (or slowed) in cells with increased (or reduced) levels of stress. The clinical relevance of understanding telomere maintenance and its interaction with biochemical stress is considerable. A growing body of evidence suggests that telomere length is linked with aging and mortality. Not only do telomeres shorten with normal aging in several tissues (e.g., lymphocytes, vascular endothelial cells, kidney, liver), but also their reduction is more marked in certain disease states. For example, there appears to be a 100-fold higher incidence of vascular dementia in people with prematurely short telomeres. Furthermore, the psychological stress associated with provision of long-term care for those with chronic illness has also been associated with premature shortening of telomeres, presumably through biochemical sequelae to the chronic activation of stress hormones. Short telomeres may therefore serve as a general indicator of previous exposure to stress and as a prognostic indicator for disease conditions in which stress plays a causative role.
An important connection between oxidative stress and aging is suggested by the accumulation of mitochondrial DNA (mtDNA) deletions and point mutations with age. Mitochondria are intracellular organelles, each carrying its own small DNA genome, which are responsible for generating cellular energy. As a by-product of energy generation, mitochondria are also the major source of ROS within the cell, and they are therefore both responsible for, and a major target of, oxidative stress. Any age-related increase in the fraction of damaged mitochondria is likely to contribute to a progressive decline in the cell and tissue capacity for energy production. Age-related increases in frequency of cytochrome c oxidase (COX)-deficient cells have been reported in human muscle and brain, associated with increased frequency of mutated mtDNA.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


