Gene
Location
Phenotype
Characteristic laboratory findings
CYP21A2
Classic forms
6p21.33
Ambiguous genitalia with virilization of females with continued postnatal virilization if undiagnosed
Normal male genitalia at birth
Acute adrenal insufficiency with salt-losing crises
Increased 17-OHP, P4, androstenedione, and ACTH
Increased PRA
CYP21A2
Nonclassic forms
6p21.33
Premature pubic hair, tall stature, irregular menses, acne, and infertility
Increased 17-OHP, P4, androstenedione, and ACTH
HSD3B2
1p12
Ambiguous genitalia with virilization of females
Ambiguous genitalia with undervirilization of male infants
Acute adrenal insufficiency with salt-losing crises
Increased 17-Preg, DHEA
Increased PRA in classic salt-losing forms
CYP11B2
8q24.3
Ambiguous genitalia with virilization of females with continued postnatal virilization if undiagnosed
Variable hypertension
Increased 11-deoxycortisol, DOC, androstenedione, and ACTH
StAR
8p11.23
Undervirilization of male infants
Acute adrenal insufficiency with salt-losing crises
All steroid hormones are low or absent
CYP17A1
10q24.3
Undervirilization of males
Delayed/absent puberty in females
Variable hypertension
Increased DOC and ACTH
Low 17α-hydroxylated steroids
Decreased PRA
POR
7q11.23
Ambiguous genitalia in males and females
Antley-Bixler skeletal anomalies, i.e., craniosynostosis, radiohumeral synostosis, midface hypoplasia, and femoral bowing
Infertility
Increased 17-OHP, P4, and ACTH
Decreased DHEA, androstenedione, testosterone
Normal electrolytes
CYP19A1
15q21.2
Virilization of female infants
Maternal virilization during pregnancy
Delayed puberty with hypogonadotropic hypogonadism and multicystic ovaries in females
Delayed/failed epiphyseal fusion
Osteopenia/osteoporosis
Impaired glucose tolerance/insulin resistance
Decreased sperm number and impaired motility
Increased androgens and P4
Increased LH and FSH
CYB5
Undervirilization of male infants because cytochrome b5 is requisite cofactor for P450c17
Decreased testosterone
Methemoglobinemia
The clinical features associated with CAH comprise a spectrum reflecting the consequences of the specific mutation. In the case of 21-OHD, the continuum ranges from salt-losing and simple virilizing forms to the milder forms. Collectively, the salt-losing and simple virilizing forms are considered to be the classic forms. The mild form is also known as the late-onset or nonclassic form (NCAH). This classification system is somewhat contrived because disease severity is better represented as a continuum based on residual enzyme activity. The incidence of the classic forms ranges from 1:5000 to 1:15,000 with variation among ethnic/racial backgrounds [1]. The prevalence of 21-OHD is lower among African-Americans than Caucasians in the United States [2]. Incomplete ascertainment muddies accurate determination of the incidence of NCAH. However, available data indicate that NCAH may occur in 1:1000 with increased frequency among Hispanics, Yugoslavs, and Ashkenazi Jews [3].
Pathophysiology
In these disorders, the loss of cortisol negative feedback inhibition leads to increased hypothalamic corticotrophin-releasing hormone (CRH) and pituitary adrenocorticotropic hormone (ACTH) secretion. The excessive ACTH secretion leads to accumulation of steroid hormone intermediates proximal to the deficient enzyme and hyperplasia of the zona fasciculata and zona reticularis.
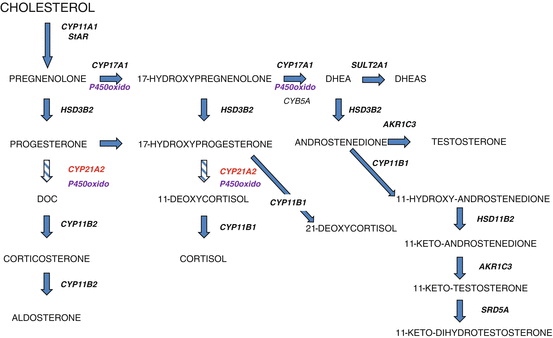
In the classical pathway of adrenal steroidogenesis (Fig. 5.1), cholesterol is converted to pregnenolone by the P450 side-chain cleavage enzyme encoded by CYP11A1. Most steroidogenic enzymes are cytochrome P450 enzymes which acquired their family name because they absorb light at 450 nm when reduced with carbon monoxide [4]. In the zona glomerulosa, pregnenolone is converted to progesterone by 3β-hydroxysteroid dehydrogenase type 2 encoded by HSD3B2. Progesterone is converted to dexoxycorticosterone by 21-hydroxylase and subsequently to aldosterone by aldosterone synthase encoded by CYP11B2. Aldosterone secretion is regulated by the renin-angiotensin system and serum potassium concentrations.
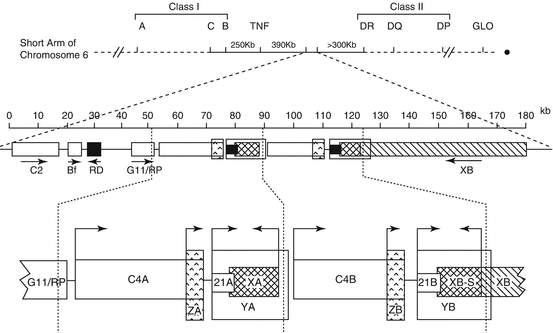
Fig. 5.1
Pathways of adrenal steroid hormone synthesis particularly relevant for 21-OHD. CYP11A1 cytochrome P450 cholesterol side-chain cleavage, StAR steroidogenic acute regulatory protein, CYP17A1 17α-hydroxylase/17,20-lyase, HSD3B2 3β-hydroxysteroid dehydrogenase type 2, P450oxido P450-oxidoreductase, CYB5A cytochrome b5, type A, SULT2A1 sulfotransferase 2A1, CYP21A2 21-hydroxylase, CYP11B1 11β-hydroxylase, CYP11B2 aldosterone synthase, AKR1C3 17β-hydroxysteroid dehydrogenase type 5, SRD5A 5α-reductase
In the zona fasciculata, pregnenolone is hydroxylated by the enzyme 17α-hydroxylase/17,20-lyase encoded by CYP17A1 to 17-hydroxypregnenolone, which is converted to 17-hydroxyprogesterone (17-OHP) by 3β-hydroxysteroid dehydrogenase type 2. Subsequently, 17-OHP is converted by 21-hydroxylase to 11-deoxycortisol, which is then converted by 11β-hydroxylase to cortisol. In the zona reticularis, the enzyme 17α-hydroxylase/17,20-lyase converts 17-hydroxypregnenolone to dehydroepiandrosterone (DHEA), which is subsequently converted to androstenedione by 3β-hydroxysteroid dehydrogenase type 2. DHEA can undergo sulfation by steroid sulfotransferase, SULT2A1, to form DHEAS.
Thus, the substrates immediately proximal to 21-hydroxylase, progesterone and 17-OHP, are elevated in patients with 21-OHD. Unfortunately, the pathophysiology of CAH is more complex than would be predicted for an autosomal recessive disorder in which the expression of the defective protein is limited to the adrenal cortex. This complexity is likely due to genetic variants at other loci which influence steroid metabolism and steroid responsiveness. More recently described alternative pathways affecting steroid hormone metabolism may also influence the clinical manifestations.
In the alternative “backdoor” pathway, 17-OHP is sequentially converted by 5α-reductase and the 3α-reductase activities of AKR1C2/4 to generate 5α-pregnane-3α,17α-diol-20-one (pdiol) that is subsequently converted to dihydrotestosterone (DHT) [5]. This alternative pathway bypasses testosterone as an intermediate. Urinary concentrations of metabolites indicative of increased flux through the alternative pathway are higher in affected individuals, particularly infants [6]. This pathway may contribute to the androgen excess responsible for prenatal virilization of affected female fetuses [7].
Under normal circumstances, the direct conversion of 17-OHP to androstenedione is not significant in humans. Yet, when 17-OHP accumulates in 21-OHD, it is metabolized by this alternative pathway [8]. Defective 21-OHD also promotes accumulation of other steroid hormone intermediates such as 21-deoxycortisol, 16α-hydroxyprogesterone, 11-ketoandrostenedione, and 11-ketotestosterone [9]. The enzyme 17β-hydroxysteroid dehydrogenase type 5 also known as aldo-keto reductase 1C3 (AKR1C3) can convert DHEA and androstenedione to androstanediol and testosterone, respectively [10]. It has been suggested that 11β-hydroxyandrostenedione, 11-ketoandrostenedione, 11β-hydroxytestosterone, and 11-ketotestosterone (11KT) are specific markers for adrenal-derived C-19 androgen hormones [11]. Regarding androgenic potency, 11β-hydroxytestosterone and 11-ketotestosterone have similar but slightly lower androgenic activity than testosterone using an in vitro cell-based luciferase reporter assay [12].
Clinical Features
Consequences of cortisol deficiency include poor cardiac function, poor vascular response to catecholamines, and increased secretion of antidiuretic hormone [13]. For 21-OHD, complete loss of function mutations abrogate aldosterone synthesis leading to hyponatremia due to impaired urinary sodium reabsorption. The hyponatremia leads to hypovolemia, elevated plasma renin levels, and, eventually, shock if not promptly recognized and treated. In the absence of aldosterone, potassium cannot be excreted efficiently resulting in hyperkalemia [14]. In 21-OHD, the elevated 17-OHP and progesterone concentrations exacerbate the mineralocorticoid deficiency because both hormones have antimineralocorticoid effects and, in vitro, interfere with aldosterone-mediated mineralocorticoid receptor transactivation [15]. In addition, the lack of prenatal cortisol exposure disrupts adrenomedullary development and can be associated with epinephrine deficiency and hypoglycemia [16].
Female infants with classical 21-OHD, either salt-losing or simple virilizing, generally present in the neonatal period with ambiguous genitalia. In some instances, the diagnosis of genital ambiguity has been suspected based on prenatal ultrasound findings. For affected female infants, the external genital findings can range from a nearly male appearance with penile urethra and bilateral undescended testes to minimal clitoromegaly. The most common physical findings in affected girls include clitoromegaly, fused rugated labia majora, and a single perineal orifice. The extent of prenatal virilization can lead to misassignment of gender at birth. Occasionally, the minimally virilized girl may not be identified until progressive clitoromegaly prompts a medical evaluation.
Affected 46,XX female infants with 21-OHD have normal female internal genitalia. The uterus can be identified on ultrasound. The ovaries may be too small to be visualized on ultrasound. Despite excessive prenatal androgen exposure, ovarian position is normal, Mullerian structures persist, and the Wolffian ducts regress. The Mullerian structures develop normally to form the fallopian tubes, uterus, and upper vagina. Virilized girls may have incomplete separation of the urethra and vagina resulting in a urogenital sinus and a single perineal orifice.
Apart from hyperpigmentation, external genital development is normal in boys with 21-OHD. Whereas girls are usually detected due to genital ambiguity, boys with salt-losing CAH appear well in the immediate newborn period. Infants with CAH tend to feed poorly and fail to regain their birth weight. Typically, they develop vomiting, hypotension, hyponatremia, and hyperkalemia in the first 10–14 days of life. Prior to implementation of newborn screening, affected boys typically presented with hyponatremic dehydration, hyperkalemia, and shock with the potential for a fatal outcome.
Pubarche refers to the development of pubic hair, axillary hair, apocrine body odor, and acne. Pubarche is the physical manifestation of adrenarche which reflects adrenal pubertal maturation and increased production of adrenal C19 steroids. Children with simple virilizing or NCAH often present with premature development of pubic hair (premature pubarche). Premature pubarche is defined as the presence of pubic hair, axillary hair, or apocrine odor developing before 8 years in girls and 9 years in boys. Additional features in children include tall stature, accelerated linear growth velocity, and advanced skeletal maturation. Clitoromegaly may develop in girls. Boys manifest phallic enlargement with prepubertal-sized testes. In a multicenter study, children less than 10 years of age most often presented with premature pubarche [17]. Among children with premature pubarche, the diagnosis of CAH should be considered when basal 17-OHP, androstenedione, and testosterone concentrations are elevated and/or bone age is advanced [18]. Nevertheless, CAH is an uncommon cause of premature adrenarche [19].
Symptoms of milder, late-onset, or nonclassic 21-OHD (NC-21-OHD) include hirsutism, irregular menses, chronic anovulation, acne, and infertility. Hirsutism, defined as excessive growth of coarse terminal hairs in androgen-dependent areas in women, has been reported to be the most common presenting feature among women [20, 21]. Hirsutism reflects the apparent sensitivity of the pilosebaceous unit/hair follicle to both circulating androgen and local androgen concentrations. Importantly, the extent of the hirsutism correlates poorly with circulating androgen concentrations [22].
Acne can occur among patients with NC-21-OHD but is rarely the primary clinical manifestation. Consideration should be given to further evaluation for patients with severe cystic acne refractory to oral antibiotics and retinoic acid treatment. Severe androgenic alopecia accompanied by marked virilization in older previously undiagnosed women has been described [23].
The nature of the symptoms leads to an ascertainment bias favoring diagnosis in affected women. Men with NC-21-OHD are typically identified through family studies. Individuals with NC-21-OHD usually do not have elevated ACTH concentrations. Some have an overresponsive ACTH-stimulated glucocorticoid response, possibly reflective of subtle adrenal hyperplasia [24].
Due to the similar clinical features, it may be difficult to distinguish women with NC-21-OHD from those with polycystic ovary syndrome (PCOS) [25, 26]. Women with NC-21-OHD tend to have higher 17-OHP and progesterone concentrations than women with PCOS [27]. Insulin resistance, obesity, polycystic ovary morphology, and elevated LH/FSH ratios tend to be more common among women with PCOS. However, none of these features clearly differentiate women with NCAH from those with PCOS [28]. Anti-Mullerian hormone concentrations do not discriminate women with NCAH from those with PCOS [29].
Family studies have demonstrated that not all individuals with genotypes consistent with NC-21-OHD develop symptoms of androgen excess. Curiously, in a study of 145 probands with 21-OHD, 4% of parents were identified to have undiagnosed or cryptic NC-21-OHD [30]. Apart from infertility among the women, these individuals had achieved normal adult heights and did not report episodes of adrenal insufficiency [30].
One uncommon feature is an adrenal myelolipoma. These adrenal mass lesions consist of myeloid, erythroid, and megakaryocytic cell lines and appear as hyperechoic masses on ultrasound and fat-containing masses on CT scan. Typically, these lesions are benign, but larger lesions are at risk for hemorrhage or rupture. MRI signal characteristics depend on the composition of the lesion.
Hypothalamic-Pituitary-Gonadal (HPG) Axis and Reproductive Concerns
Oligo-amenorrhea , chronic anovulation , and infertility are common presenting complaints for women with NC-21-OHD and can occur in women with classic 21-OHD despite adequate hormone replacement therapy. Hence, women with 21-OHD can develop a secondary “PCOS” phenotype [31]. The specific molecular mechanisms responsible for the altered hypothalamic-pituitary-adrenal (HPO) axis function accompanied by apparent ovarian androgen excess are unclear. Increased circulating concentrations of adrenal androgens and progestins likely influence HPO axis function.
Additional features affecting female reproduction include vaginal stenosis with dyspareunia , impaired sensation , changes in cervical mucous, poor self-esteem, and disinterest in having children. Impaired quality of life and risk for depression have been reported to be higher in women with 21-OHD [32]. Potential contributing factors include engaging in high-risk behaviors, perception of being different from other women, and perceived lack of autonomy [33]. Reproductive outcomes for women with CAH can be greatly improved by adequate suppression of progesterone and 17-OHP to promote ovulation and implantation of the fertilized ovum; this may require optimizing both glucocorticoid and mineralocorticoid therapies [34]. Occurrence of miscarriages is higher among untreated women with NC-21-OHD [20, 21]. The consequences of prenatal androgen exposure on the developing female brain are being explored [35].
Quality of life (QoL) for women with CAH has been a long-standing concern for women with CAH. One series reported later sexual debut, fewer pregnancies and children, and increased incidence of homosexuality; these outcome measures were related to type of surgical correction and the severity of their mutations [36]. Girls with CAH are reported to prefer more masculine toys, more male-dominant occupations, rougher sports, and non-heterosexual orientation [37]. Another series of 24 women, who answered a questionnaire, reported that 87.5% of women indicated that CAH had not interfered with their social relationships [38]. Regarding gender identity and sexual orientation, 25% of women indicated that they had occasionally wished to be a man, and 62% reported having heterosexual orientation at all times [38]. This area of investigation has been confounded with several factors including small numbers of subjects, lack of control subjects, changes in surgical techniques over time, and variability of age at the time of surgical correction.
Careful consideration regarding surgery is urged for girls with genital ambiguity. Only experienced surgeons/urologists should perform feminizing genitoplasty and vaginal reconstruction [39]. During adolescence, the adequacy of the vaginal introitus for the use of tampons and sexual intercourse should be assessed. For girls with vaginal stenosis, dilatation is often helpful.
Gonadal adrenal rest tumors , predominantly testicular adrenal rests (TARTs) , occur in up to half of men with 21-OHD. These tumors arise from adrenal cells that descend with the testes during testicular development. TARTs are not malignant but can compress the rete testis and seminiferous tubules culminating in testicular atrophy and obstructive azoospermia. Ultrasound and MRI are helpful to detect TARTs less than 2 cm because small lesions are generally not palpable. TARTs may be present in childhood and adolescence and have rarely been described in men with NC-21-OHD [40, 41]. Although TARTs have been attributed to poor adherence with hormone replacement therapy, pathogenesis of TARTS may be more complicated [42]. In affected males, the elevated adrenal C19 steroid secretion can suppress gonadotropin secretion resulting in hypogonadotropic hypogonadism and subsequent oligospermia. Ovarian adrenal rest tumors (OARTs) have been infrequently reported in affected women.
Molecular Genetics
The CYP21A2 gene is located in a complex genetic region at chromosome 6p21.3 where it lies in close proximity to a highly homologous pseudogene, CYP21A1P. CYP21A2 and CYP21A1P are arranged in tandem repeats with the C4A and C4B genes, which encode complement component 4. The tenascin (TNX) and serine threonine nuclear protein kinase (RP) genes are also mapped to this region. These four genes, RP, C4, CYP21, and TNX, form a unit known as RCCX. Most alleles carry two RCCX units in which one has CYP21A2 and the other has CYP21A1P (Fig. 5.2
).
Fig. 5.2
Genetic organization of CYP21A2 and CYP21A1P. This figure illustrates the location of the C4A, CYP21A2, C4B, and CYP21A1P genes on the short arm of chromosome 6
To date, over 200 CYP21A2 mutations have been reported (http://www.hgmd.cf.ac.uk; www.cypalleles.ki.se). Yet, despite the large number of reported mutations, approximately ten mutations account for the majority of affected alleles. Most mutations result from gene conversion events in which the functional gene acquires deleterious CYP21A1P sequences or from misalignment during meiosis that can give rise to duplication or deletions of the RCCX unit. Haplotypes with three or four RCCX units have been described [43]. Another example of misalignment is a CYP21A1P/CYP21A2 chimera in which a portion of the CYP21A1P gene is fused to a portion of the CYP21A2 gene [44]. Rarely, CAH can be associated with uniparental disomy [45]. The de novo mutation rate is approximately 1%.
Most affected individuals are compound heterozygotes with different mutations on each allele. Mutations range from complete loss of function to mild missense mutations. Estimates of in vitro 21-hydroxylase activity range from <1% for mutations associated with salt-losing CAH to 2–10% for simple virilizing CAH and to 30–50% for NCAH. Genotype, residual enzyme activity, and phenotype generally correlate such that an individual’s phenotype reflects their milder mutation. Patients with classical salt-losing CAH usually carry complete loss of function mutations on both alleles. Patients with simple virilizing CAH typically have a complete loss of function mutation on one allele and the I172N or intron 2 splicing mutation on their other allele. Patients with NCAH often carry different mutations with at least one allele carrying a mild missense mutation such as V281 L. Approximately 25–50% of individuals with NCAH are reported to have mild mutations on both alleles [46–48]. Mutations associated with NCAH include V281L, P453S, and R339H. The P30L mutation is often detected in patients with NCAH but is typically associated with more severe androgen excess [49].
As noted above, the CYP21A2 locus is quite complex which precludes molecular genetic analysis as the first-line diagnostic test. Molecular genetic testing is also confounded by the possibility of multiple mutations on a single allele and the presence of different CYP21A2 mutations in one family. Multiple genetic testing strategies such as PCR-based mutation detection methods, sequencing, and multiplex ligation-dependent probe amplification may be needed to accurately interrogate and segregate the mutations in an affected individual. In some instances, it may be necessary to perform genetic analyses on the parents to segregate the specific maternal and paternal mutations and confirm that mutations are on opposite alleles. Despite these potential obstacles, genetic analysis can be a useful adjunct to newborn screening [50].
As noted above, one RCCX unit contains CYP21 and TNX genes (Fig. 5.2). TNXB encodes tenascin-X, which is an extracellular matrix glycoprotein involved in collagen organization and matrix integrity. Mutations in TNXB are associated with Ehlers-Danlos syndrome. Several distinct alleles have been characterized with loss of CYP21A2 and specific TNXB alleles. Patients have been described to have monoallelic or biallelic TNXB variants. The severity of the Ehlers-Danlos syndrome is dependent on the TNXB genotype. Patients with CAH will benefit from evaluation for features associated with Ehlers-Danlos syndrome such as hypermobile joints and skin laxity [51].
Diagnosis
An elevated 17-OHP concentration provides confirmation of the diagnosis of 21-OHD deficiency. Most affected infants have random 17-OHP values >5000 ng/dl (150 nmol/L) [52]. For infants, additional laboratory evaluation can include electrolytes, plasma renin activity, progesterone, and androstenedione concentrations. Pelvic ultrasound imaging and chromosome analyses are recommended for virilized female infants.
For individuals with symptoms suggestive of NC-21-OHD, an early morning basal 17-OHP has been suggested as an effective screening test. Armengaud et al. reported 100% sensitivity and 99% specificity with a threshold value of 200 ng/dl (6 nmol/L) to diagnose NC-21-OHD in children with premature pubarche [19]. A bone age X-ray should be obtained to assess for acceleration of skeletal maturation.
Blood samples for 17-OHP determinations should be obtained in the follicular phase for reproductive-aged cycling women because the 17-OHP concentration may be elevated during the luteal phase. In this situation, Escobar-Morreale et al. recommended using a basal 17-OHP of 170 ng/dl (5.1 nmol/L) as the “cut point” for women [53]. Nevertheless, for any age group, an ACTH stimulation test may be warranted to complete the evaluation for 21-OHD. For an ACTH stimulation test, following collection of a basal blood sample, 0.25 mg synthetic ACTH (Cortrosyn) is administered by intravenous or intramuscular routes; a second blood sample is collected at 30 and/or 60 min. In addition to 17-OHP, cortisol should be measured especially among individuals with NC-21-OHD to assess the adequacy of cortisol secretion. To differentiate 21-OHD from other disorders of steroidogenesis, determination of progesterone, 17-hydroxypregnenolone, 11-deoxycortisol, DHEA, deoxycorticosterone, and androstenedione may be warranted [54].
In general, CYP21A2 mutations on both alleles will be identified when ACTH-stimulated 17-OHP concentrations are greater than 1500 ng/dl (45 nmol/L). However, some individuals with diagnostic genotypes have ACTH-stimulated 17-OHP values between 1000 and 1400 ng/dl (30–45 nmol/L). Individuals with 21-OHD have elevated 21-deoxycortisol concentrations, but commercial availability of this hormone assay is limited. Liquid chromatography-tandem mass spectroscopy (LC-MS/MS) has demonstrated elevated 17-OHP, 21-deoxycortisol, 16α-hydroxyprogesterone, and progesterone; these steroids comprise sensitive and specific biomarkers to accurately identify patients with CAH due to 21OHD [9]. As noted above, the 11oxo-C19 steroids, 11β-hydroxyandrostenedione, 11-ketoandrostenedione, 11β-hydroxytestosterone, and 11-ketotestosterone (11KT), are elevated in 21-OHD [11].
Related posts:
 Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Zonation and Development
Adrenal Zonation and Development
 Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
Diagnosis and Management of Adrenal Insufficiency
Pheochromocytomas and Paragangliomas: Genetics and Pathophysiology
 Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
Adrenal Cushing’s Syndrome: Updates on Overt and Mild Hypercortisolism
 Diagnosis and Management of Adrenal Insufficiency
Diagnosis and Management of Adrenal Insufficiency
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


