Using immunohistochemistry, the cell types of these immune infiltrates can be defined: quantitatively, the most abundant immune cells are CD3+ lymphocytes (mostly composed of CD4+ T-helper cells, CD8+ cytotoxic T cells, and FOXP3+ regulatory T cells). Also, CD68+ and/or CD163+ macrophages are extremely abundant in many solid tumors. Enumeration of these cells, for example, by means of digital pathology, has proven to be effective in predicting survival, chemotherapy response, and immunotherapy response [11–13]. Additional to enumeration of cells in the invasive margin and the tumor core, other spatial analysis methods enable the definition of higher-order spatial features of immune cell infiltrates: continuous approaches for density analysis (Fig. 13.2, center) do not rely on predefined static regions, but enable a more fine-grained analysis of spatial patterns, including spatial clustering. Multivariate approaches (Fig. 13.2, right) enable the analysis of co-localization of different markers, thereby allowing to generate hypotheses about mechanistic interaction between different immune cell subtypes.
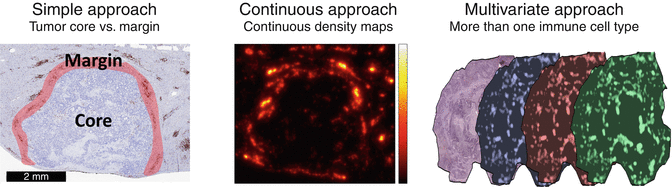
Fig. 13.2
Methodical approaches to quantification of immune cell infiltrates. Left: a standard method is to delineate a “core” region and a “margin” region and to count cells separately in each of these regions. Middle: cells can also be analyzed in a continuous way, enabling detailed analysis of spatial clustering. Right: Integrating multiple spatially aligned staining can yield information about co-localization of different immune cell subtypes
Interestingly, spatial patterns of immune infiltrates within or close to the tumor tissue only reflect one part of the host response to the tumor. It has been shown that host response can also be detected at a distance, most notably in “tertiary lymphoid structures” that emerge at a distance of up to several millimeters around solid tumors. These features are most notable in lung cancer and have also been detected in other entities such as colorectal cancer [14–16].
13.3 Genetic Characteristics of the Tumor Influencing the Host’s Immune Response
Cancer is a genetic disease as the accumulation of several driver mutations eventually transforms a cell in a cancer cells. In general, the immune system identifies most cells with genetic aberrations and eliminates them in order to prevent the occurrence of malignant transformation. However, certain cancer-initiating cells manage to overcome this initial immune elimination and enter a process of equilibrium. Consequently, the selection pressure for cells able to evade the immune response rises, and eventually they escape the immune regulatory mechanisms [2]. Although these cancer cells have theoretically evaded the immune system, they remain an immunogenic target as the infiltration with immune cells in established cancer samples proofs [11, 14, 17].
Molecular cancer subgroups based on the presence of certain driver mutations were shown to correlate with characteristics of the inflammatory tumor microenvironment. Interestingly, several known targetable driver mutations, like EGFR mutation, estrogen overexpression, or presence of IDH mutation, are rather associated with a so-called “cold” inflammatory tumor microenvironment and subsequently rather low density of tumor-infiltrating lymphocytes and absence of programmed cell death ligand 1 (PD-L1) expression [18, 19]. Further, loss of PTEN was shown to enhance immune escape as increased expression of immunosuppressive cytokines results in reduced T cell-mediated tumor killing and T cell trafficking into the tumor. In line, application of a PI3Kβ Inhibitor reduced these immunosuppressive properties in preclinical models and as a consequence the efficacy of anti-PD1 and anti-CTLA4 treatments [20].
The genetic aberrations of a tumor are further the most important immunological target, as the immune system can detect peptides generated from aberrantly expressed genes or neo-antigens expressed as a direct consequence of somatic mutations. The resulting proteins are divergent from the germ line sequence and therefore entirely absent in the normal human genome. Importantly, while driver mutations directing malignant transformation are similar in a given tumor histology, the specific mutation signature and consequently the (neo-)antigen composure of any particular tumor is largely distinct, resulting in highly diverse (neo-)antigen signatures in tumors of the same histology [21]. These (neo-)antigens are displayed on the major histocompatibility complexes (MHCs; defined by the human leukocyte antigen (HLA) haplotype) on the surface of malignant cells and can thereby be identified by T cells, which are able to generate a tumor-specific cytotoxic immune reaction. However, the presence of (neo-)antigens does not equal a meaningful tumor-specific T cell reactivity [22]. Abnormalities of the MHCs in cancer cells including loss or mutations of genes encoding the MHC heavy chains as well as epigenetic modifications may impair the recognition of tumor cells by the immune system [23]. Furthermore, the presence of (neo-)antigens throughout a given tumor is not a static condition as the genomic landscape of a tumor is heterogeneous in space as well as in time. Spatial genetic heterogeneity is especially observed within a tumor bulk and at least partly explained by cell migration on a small scale [24]. In line, the immunogenicity of a tumor differs locally, resulting in a heterogeneous infiltration with immune cells as observed in various tumor entities. Immunosuppressive factors, like programmed cell death ligand 1 (PD-L1), frequently show a heterogeneous expression throughout a given tumor [25]. PDL1 expression in only 1% of tumor cells might already substantially impact the immunogenicity and is associated with survival prognosis in entities like breast cancer and lung cancer [26, 27]. In conclusion this data suggests that the spatially heterogeneous genetic landscape of tumors shapes the also spatially heterogeneous immune response.
Besides this spatial heterogeneity, the genetic characteristics of a given tumor change over time. Here, matched primary tumor and metastatic samples revealed marked genetic differences in a process called branched evolution from the common ancestor cell to the primary tumor and the metastasis [28]. As a result, also the (neo-)antigen signature is not a static condition over time: first, due to the genetic evolution of the tumor itself and second due to immune editing as response to survival advantage of non-immunogenic cancer cell clones [2, 29].
13.4 Host-Specific Genetic Characteristics Determining the Tumor-Specific Immune Response
Importantly, besides the above outlined genetic characteristics of the tumor, also the characteristics of the host’s immune system determine the efficacy of the resulting immune response. Density of tumor-infiltrating lymphocytes and more precisely preexisting CD8+ T cells in the invasive tumor margin are predictive markers for the response to PD1 checkpoint inhibitors as well as chemotherapy [13, 30]. However, the magnitude of T cell infiltration and the ratio of T cell subsets vary substantially between patients, despite tumor histology and location being similar. In line, identical to the genomic evolution of the tumor cells, also the T cell infiltration phenotype might underline a host-specific evolution generating the observed heterogeneity in tumor cell genotype and immune cell infiltration (Fig. 13.3).
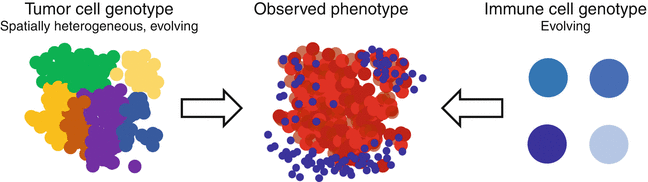
Fig. 13.3
Immune infiltrates in solid tumors typically show a spatial and temporal heterogeneity. This heterogeneity is driven by tumor factors (e.g., genetic heterogeneity) and host factors (e.g., adaptive immune response)
The host factors’ impact on the tumor-specific T cell response has been only partly understood and so far less intensively investigated compared to tumor-specific characteristics. One important factor here might be T cell receptor (TCR) repertoire, which is unique in every person as the TCR-β alleles are subjected to strict allelic exclusion. The TCR repertoire expressed in tumor-infiltrating T cells determines the degree of tumor reactivity and (neo-)antigen specificity, and a further insight could potentially function as predictive signature for immune modulating therapies [31]. The TCR clonality of the host is an important predictive factor for the response to PD1 checkpoint inhibitors [32].
Furthermore, adaptive immune responses within a certain organ might impact the host’s immune cell phenotype. Organs with a comparably strict immune regulation, such as the brain, might actively restrict the generation of an active immune response. In line, the density of tumor-infiltrating lymphocytes in primary brain tumors is rather low and matched brain metastases present with a less active inflammatory tumor microenvironment compared to their matched primary lung cancer samples [33, 34].
The HLA haplotype is a further host-specific factor determining the anticancer immune response, and it defines the binding capacity with a certain (neo-)antigen and as a consequence the resulting T cell-mediated immune response. In line, vaccination strategies using specific, single neo-antigens are frequently restricted to patients with a certain HLA haplotype as only this certain combination has the potential to induce a specific immune response [35, 36]. A recently upcoming host factor influencing the cancer-specific immune response is the gut microbiome, and further studies concentrate on their interference as well as the therapeutic implications [37].
Further systemic host characteristics shaping the intensity of the tumor-specific immune response are circulating inflammatory cytokines like interleukin-6 that are associated with a chronic inflammatory state in cancer patients and induce cancer-associated cachexia [38]. In line, blood markers indicating chronic inflammation like increased C-reactive protein (CRP), increased white blood cell counts, or microcytic anemia are associated with impaired survival prognosis in several cancer types [39, 40]. High concentration of tumor necrosis factor alpha, interferon gamma, as well as the chemokine ligands CCL2, CCL4, or CXCL10 in the blood circulation might be associated with an increased tumor-supporting activity of circulating myeloid-derived suppressor cells [41, 42]. Liquid biomarkers like plasma level of soluble PDL1 were shown to be associated with poor prognosis and suppression of antitumor immunity in patients with advanced lung cancer [43]. In conclusion, not only specific factors of the tumor but rather the very personal interaction of a specific tumor and a specific host immune system determine the amplitude of an antitumor immune response.
13.5 Tumor-Specific Genetic Alterations as a Target for Vaccination
In contrast to more general immune activating therapies like immune checkpoint inhibitors or interferons, vaccination induces a more specifically directed immune response to distinct tumor epitopes [44]. Here, several techniques including active vaccination, meaning application of vital, active immune cells like dendritic cells, CAR T cells, or neo-epitope-specific T cells, or passive vaccination with peptides or RNA/DNA constructs either against one or multiple tumor-specific epitope(s) are applied (Table 13.1).
Table 13.1
Vaccination approaches
Vaccination approach | Principle | Challenges |
|---|---|---|
Mono neo-antigen | (Neo-)antigen vaccination of a frequently expressed peptide or RNA/DNA | Heterogeneity of antigen presence; selection pressure for cells without antigen; HLA haplotypes |
Multi-neo-antigen | Vaccination of several frequently expressed (neo-)antigens | Heterogeneity of antigen presence; selection pressure for cells without antigen; HLA haplotypes |
Mutanome specific | Identification of patients’ specific (neo-)antigens and formulation of specific multi-neo-antigen vaccination | Identification of antigens with immunogenic properties; complex production |
Dendritic cell vaccination | Ex vivo activation of patient-derived dendritic cell to increase antigen presentation | Activation with mono (neo-)antigen or tumor-specific lysate; complex production |
Adoptive transfer of neo-epitope-specific T cells | Ex vivo expansion of tumor-infiltrating lymphocytes specifically reactive a certain (neo-)antigen | Complex production |
CAR T cells | Genetically engineering of patient-derived T cells in order to express an antigen-specific T cell receptor | Heterogeneity of (neo-antigen) presence; selection pressure for cells without (neo-)antigen; complex production |
Active immunotherapy approaches use patient-derived cells, activate and expand them ex vivo, and transfer them back in the patient. This personalized immunotherapy requires demanding and complex methods, complicating the commercialization and conduction of large phase III clinical trials [44, 45]. Dendritic cell vaccination is an approved therapeutic approach, which has shown to be safe and generate responses, e.g., in melanoma, glioblastoma, and prostate cancer [46–48]. In brief, peripheral monocyte and dendritic precursor cells are taken from the patients and amplified ex vivo. Next, the monocytic cells are matured using an activation cocktail containing pro-inflammatory cytokines, CD40 ligand, or TLR agonist. The matured dendritic cells are then loaded with tumor antigen either by inoculation with peptides, proteins or known tumor neo-antigens or with patients’ specific tumor lysates [44, 49]. These activated, matured dendritic cells are reinfused in the patients and can induce a specific immune response by antigen presentation to T cells as well as support of B cell memory maintenance and activation of natural killer cells [47, 49]. So far, dendritic cell vaccination therapy has only showed a survival benefit in one prospective phase III study in metastatic castration-resistant prostate cancer [48].
Adoptive transfer of neo-antigen-specific T cells is another immunotherapy approach currently investigated in clinical trials and was shown to generate immune response also in tumor entities that are generally not prone to immune modulating therapies, like colorectal cancer and cholangiocarcinoma [50, 51]. Here, next-generation whole genome sequencing is used to reveal non-synonymous mutations potentially functioning as neo-epitopes. Dendritic antigen-presenting cells, retrieved from the specific patient, are then loaded in vitro to present these neo-epitopes to T cells isolated from the tumor tissue (tumor-infiltrating lymphocytes). T cells responding specifically to the presented neo-antigen are then expanded ex vivo and subsequently reinfused into the patient. Here, the neo-antigen-specific T cell can mount a tumor-specific immune response, resulting in the persisting presence of these antigen-specific T cells and potential tumor control [50, 51].
Another immunotherapy approach is the application of T cells with a chimeric antigen receptor (CAR) [44]. To generate an antigen-specific CAR T cell type, peripheral T cells are harvested from a patient. Next, the desired antigen-specific CAR is introduced in the T cells trough viral or nonviral methods resulting in T cells with genetically engineered, antigen-specific T cell receptor. The CAR T cells are expanded ex vivo and reintroduced in the patient [52, 53]. Here, CAR T cells can target cells expressing the antigen and enhance T cell effector function in an MHC-independent manner. Therefore, the HLA haplotype potentially does not affect the efficacy of CAR T cell therapy [52]. Neo-antigens like human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), mesothelin, and carcinoembryonic antigen (CEA) among others are currently tested as potential targets for CAR T cells in solid cancers [53–56]. Besides the already substantial clinical efficacy in hematologic malignancies, the first case reports also indicate efficacy in solid cancers and several studies are currently recruiting [54, 56].
Besides treatment approaches using ex vivo cultured patient-specific cells, several peptide- or RNA/DNA-based vaccination approaches have been postulated in order to increase the host’s immune response by increasing the availability of (neo-)antigen(s). Mono neo-epitope vaccinations encompass one specific antigen of a given tumor. The targeted epitope has to be expressed in the majority of cancer cells to ensure that the initiated T cell response is effective. Clinical trials investigated, e.g., a HER2-specific vaccination in HER2 overexpressing breast cancer or gp100 vaccination in melanoma [57, 58]. However, the mono epitope-directed immune response results in a selection pressure of the antigen-positive cancer cells and provides a survival advantage to negative cells, potentially resulting in an immune escape. Furthermore, the affinity of the peptides to the MHC varies according to the HLA haplotype, restricting the efficacy of certain peptide vaccines to patients expressing a specific HLA haplotype [59].
A multi-epitope vaccination using several frequently present epitopes of a tumor can potentially generate a broader immune response and prevent immune escape more efficiently compared to mono epitope vaccination. A set of predefined epitopes, which have been identified to be frequently expressed in a certain tumor type, are included in the vaccination [60, 61]. However, as outlined before, the specific antigen signature of a certain patient is very unique although histology and location of the given tumors might be comparable [62]. Mutanome-specific vaccination provides an even more personalized vaccination approach: specific multiple neo-epitopes can be identified in a given patient and facilitate the manufacturing of an individual vaccine [63]. These neo-epitopes can be identified either via genome sequencing and in silico antigen prediction or directly via mass spectrometry [63, 64]. Hereby, tumor heterogeneity is more adequately addressed, and clinical application is more broadly possible, as limitations like the presence of a certain antigen or HLA haplotype are reduced [64]. However, not all genetic alterations result in transcribed proteins and second in the generation of immunogenic epitopes. Only about 20–40% of (neo-)antigens are recognized by the immune system and result in the generation of a tumor-specific T cell response [64]. Therefore, a key question is how immunogenic cancer mutations of relevance can be identified and therapeutically explored in a personalized manner. Here, the numerical difference NetMHC score between the wild-type peptide and the mutated neo-antigen and the conformational stability of the MHC I and peptide interaction are essential to define the immunogenicity of a given neo-antigen [65].
Currently, several clinical trials are testing various neo-epitope vaccine approaches, either in monotherapy or combination. An important matter to discuss, as for the application for immune checkpoint inhibitors, is to identify which patients will most likely benefit from these specific vaccination approaches. Here, the extend of the disease should be taken into account. Vaccination, although generating a tumor-specific immune response, does likely not result in tumor shrinkage but rather control of existing tumor. Therefore, patients with minimal residual disease are likely to benefit most. Therefore, careful trial design is essential to identify patients benefiting most from vaccination-based treatment approaches.
13.6 Genetic Alterations of the Tumor as Predictive Biomarkers for Immune Response to Immune Checkpoint Inhibitors
Besides the high response rates observed in patients treated with CTLA4 and PD1 axis targeting immune checkpoint inhibitors, a significant fraction of patients does not respond. Therefore, accurate and precise predictive biomarkers are urgently needed to adequately select patients with the highest likelihood of response (Table 13.2).
Table 13.2
Genetic alterations of the tumor as predictive biomarkers for immune checkpoint inhibitor based therapy
Predictive marker | Immune modulating therapy | Entity | References |
|---|---|---|---|
Number of somatic mutations | CTLA4 and PD1 blockade | Melanoma, non-small cell lung cancer | |
Mismatch repair deficiency | PD1 blockade | Colorectal cancer | [66] |
Neo-antigen signature/load | PD1 and CTLA4 blockade | Lung adenocarcinoma, melanoma | |
Copy number loss | CTLA4 blockade | Melanoma | [32] |
Indeed, rather the total mutational load than the presence of specific alterations was shown to correlate with an immune response and increased likelihood of response to immune checkpoint inhibitors (Fig. 13.4) [7, 68]. In line, cancer types with high mutational load like melanoma or lung adenocarcinoma are more sensitive to immune modulating therapies in comparison to tumors with less frequent mutations like, e.g., breast cancer [63]. Cancer types induced by a high carcinogenic exposure like melanoma trough UV radiation or lung cancer in smokers, or virally induced cancer types such as head and neck squamous cell carcinoma present with a particularly high mutational burden [69]. Interestingly, the mutational load differs greatly between the individual cancers, although other characteristics like histology, location, and presence of driver mutations are very similar [8]. Some cancer types with a rather low mutational burden, like colorectal cancer or glioblastoma, might respond impressively in the presence of certain specific genetic subtypes like the microsatellite instability or mismatch repair deficiency [66, 70, 71]. However, also cancer types with a lower median number of somatic mutations have shown impressive and durable responses like renal cell carcinoma, arguing that besides the raw number of mutations, the immunological quality of the mutations might influence the cancer immunosurveillance. More precisely, the mutational load correlates with immunogenicity of a given tumor as each additional mutation increases the odds that a relevant patient-specific cancer neo-antigen is created.
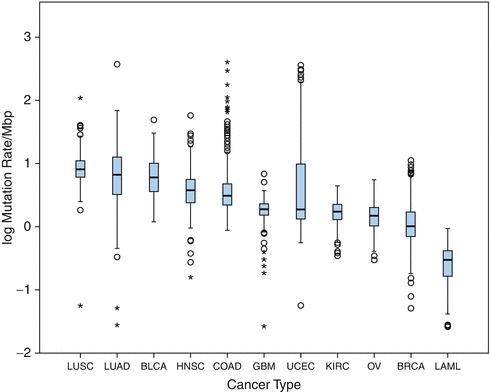
Fig. 13.4
Mutational load of the different primary tumor types, adapted from [69]; LUSC lung squamous cell carcinoma, LUAD lung adenocarcinoma, BLCA bladder urothelial carcinoma, HNSC head and neck squamous cell carcinoma, COAD colon adenocarcinoma, GBM glioblastoma, UCEC uterine corpus endometrial carcinoma, KIRC kidney renal clear cell carcinoma, OV ovarian serous cystadenocarcinoma, BRCA breast invasive carcinoma, LAML acute myeloid leukemia
Indeed, a high number of mutations and a high load of neo-antigens are associated with increased infiltration of CD8+ tumor-infiltrating lymphocytes and improved prognosis [62, 72]. This neo-antigen signature can vary in its immunogenicity depending on the affinity to the MHC complex, the difference to wild-type peptides, and the heterogeneity of the neo-antigen within the tumor. The majority of mutations found in melanoma and other tumors with a high mutational load are “passenger mutations” that are unrelated to the cellular transformation process. As such, the vast majority of potential neo-epitopes in these cancers are patient-specific. In melanoma patients responding to CTLA4 inhibitors, a specific neo-antigen signature of neo-epitopes homologous to many viral and bacterial antigens was discovered [9]. Whether this particular neo-antigen signature also has predictive potential in other entities as well as the definitive cutoff values defining a tumor with high mutational load remains unknown [8, 9].
Furthermore, the burden of copy number loss was shown to correlate with likelihood of response to CTLA4 inhibition. Here, copy number loss is associated with melanoma progression and might therefore be one cause of immune escape as a correlation of copy number loss and downregulation of immune-related gene expression was found [32]. Interestingly, effects of low copy number loss and high mutational load had nonredundant effects on the clinical response, indicating that the combination of several predictive markers could potentially provide a more precise prediction of response [32]. Some truncating genetic alterations encoding the interferon-receptor-associated Janus kinases 1 (JAK1) and 2 (JAK2) were shown to correlate with acquired resistance to anti-PD1 immune checkpoint inhibitors [29].
13.7 Personalized Immunotherapy: Combination of Immune Modulation Therapies
Although encouraging response rates have been observed with immune modulating therapies, the majority of patients still will not benefit in the long term from currently available treatments. This might be due to several simultaneously active immune escaping factors of the tumor in addition to the host-specific factors. Combination of immune modulation therapies with each other or with other established treatment options can enhance the interaction of host and cancer by improving different steps of the cancer-immune cycle [4].
A lack of available antigen can be overcome by vaccination with tumor-specific antigens. Indeed, preclinical studies suggest a synergistic effect of vaccination with checkpoint inhibition, although a clinical study investigating the combination of CTLA4 inhibition and gp100 vaccination did not show a clinically meaningful additional efficacy [57, 73]. However, the recent insights on peptide vaccination therapies including the establishment of mutanome-specific vaccinations might provide a better combination partner for immune checkpoint inhibitors.
The so-called abscopal effect argues for the reasonable combination of radiotherapy and immune checkpoint inhibitors [74]. In theory, radiotherapy first increases the availability of antigens by apoptosis of tumor cells inducing tumor-specific T cell responses. In addition, radiotherapy has several local effects on the inflammatory microenvironment and induces the infiltration of the tumor with antigen-presenting cells, macrophages, and cytotoxic T cells [10, 75]. Therefore, vaccination and radiation but also other therapies resulting in increased antigen presentation can overcome insufficient antigen transport, and presentation and combination with immune checkpoint inhibitors might be synergistically [76, 77]. However, today little is known about how to identify patients with insufficient antigen presentation, as no reliable markers to quantify efficacy of antigen presentation have been proven.
As outlined above, a tumor-specific immune response is highly regulated as several TCR co-receptors control the resulting T cell response with either enhancing or suppressive signals [78]. The PD1 and CTLA4 axis are both T cell response suppressing pathways, actually reducing the activation of T cells. In line, the inhibition results in an “unleashing” of the tumor-specific T cell response [78]. However, PD1 checkpoint inhibition results in upregulation of other immunosuppressive immune checkpoint pathways like TIM3 [59]. Therefore, combination of two checkpoint inhibitors might overcome this immune escape mechanism. The combination of CTLA4 and PD1 blockade was shown to be more clinically effective compared to either checkpoint inhibitor alone, although in light of a higher toxicity rate [79, 80]. Further, CTLA4 therapy was shown to increase TCR clonality present in the tumor and thereby enhance the likelihood of response to PD1-directed therapies [32]. Several studies are currently investigating the clinical efficacy of second-generation checkpoint inhibitors targeting, e.g., TIM3, LAG3, OX40, and others in combination with first-generation checkpoint inhibitors.
Further combinations aim to increase the intra-tumoral T cell density in addition to an immune checkpoint inhibitor. The PI3K and MEK pathways were shown to influence the density of T cell within the tumor tissue [20, 81]. Addition of MEK tyrosine kinase inhibitors to immune checkpoint inhibitors might therefore result in increased density to tumor-infiltrating lymphocytes, thereby converting “cold” tumors into “hot” ones, which are then more susceptible for immune checkpoint inhibitor therapy [82].
A further promising combination is the addition of immune checkpoint inhibitors to standard chemotherapy regimes, as many standard chemotherapies have been shown to have immune modulating function [44]. The response rate as well as the progression-free survival was increased significantly by the combination of standard platinum-based chemotherapy and a PD1 inhibitor [83, 84].
Lastly, in order to generate a durable adaptive immune response, the innate immune system has to be considered as well. It has long been known that macrophages and neutrophils modulate T cell activity in a clinically relevant way [85]. Recently, it was shown that macrophage repolarization can lead to an increased lymphocyte response against the tumor in otherwise non-immunogenic tumors [86, 87]. Currently, clinical trials exploiting these regulatory axes are ongoing.
In summary, cancer immunotherapy is rapidly changing from single-agent approaches toward combination therapies addressing the patient-specific interactions between the immune system and the given cancer [88]. The combination of immune modulating therapies certainly creates another layer of complexity that has to be addressed by better preclinical understanding of the underlying biology and resulting reasonable biomarkers. Here, several genetic properties of the tumor itself as outlined above but also host-specific factors and dynamic factors caused by the interaction of specific host and tumor have to be incorporated in potential predictive algorithms. In depth characterization of the inflammatory tumor microenvironment, genetic characteristics of the tumor and the host have to be assessed and further analyzed to develop predictive biomarker signatures. We have to get used to monitor immediate treatment effects in sequential tumor biopsies to better understand the mechanisms of response, even more important to understand the mechanisms of resistance in the individual patient. Such data will be very important to feed our prediction algorithms. Ultimately, we can envision clinical trial strategies where every patient is receiving a different drug combination that might even change in sequence. Real-time assessment of treatment effects on the tumor will help to constantly adopt and modify the treatment strategy in the individual patient.
References
1.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45. doi:10.1016/S0140-6736(00)04046-0.CrossrefPubMed
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


