5 Genetics
Introduction
Most breast cancers are sporadic and likely due to a combination of genetic and environmental factors, but an estimated 5% to 10% of breast cancers are “hereditary,” meaning attributable primarily to mutations in a specific gene. About 50% of hereditary breast cancers are believed to be due to the hereditary breast and ovarian cancer syndrome (HBOC).1,2 With only 1% of breast cancers being attributable to a handful of very rare syndromes (discussed in the text that follows), this leaves 50% that are due to yet undiscovered genetic factors.
Recognizing hereditary breast cancer is an academic exercise, but it can also be helpful to families for several reasons. For a woman with breast cancer, the identification of a mutation can help to explain her cancer as well as predict her risk of developing other related cancers. For a woman with no personal history of cancer, but with a strong family history of breast cancer, it can help to better define her risk of breast and other related cancers, enabling consideration of more specific screening and risk reduction options. In both cases, such genetic information can also be useful when assessing risk to family members, particularly children, siblings, and parents. Such information may also decrease anxiety, since studies have shown that women with a family history on average have a higher perceived risk of developing breast cancer than their actual risk.3
Hereditary Breast and Ovarian Cancer Syndrome (HBOC)
Features and Cancer Risks
The two genes BRCA1 and BRCA2 were initially discovered by studies of families strongly suggestive of hereditary breast cancer. Thus, they are named breast cancer susceptibility gene 1 (BRCA1) and breast cancer susceptibility gene 2 (BRCA2). Several hundred mutations have been reported in BRCA1 and BRCA2 since their identification, some of which are associated with an increased cancer risk.4
Functions of BRCA Genes
The involvement of the BRCA1/2 genes in DNA repair is also thought to explain why women with BRCA1/2-related breast cancer have a better response to cisplatin-based regimens than those with sporadic breast cancer.5 Cisplatin generates a highly reactive species after intracellular aquation. This species binds to DNA, causing intra-strand cross-links primarily between adjacent guanines in the DNA helix major groove. Among other cytotoxic effects, it is believed that the cisplatin-induced adducts may induce the mismatch repair (MMR) complex to produce single-stranded DNA breaks, resulting in cytotoxicity and cell death.6
Prevalence and Penetrance of BRCA1/2 Mutations
For genetic testing of BRCA1 and BRCA2 to be useful to families at high risk for a mutation in these genes, it is necessary to have accurate assessments of their prevalence (how common the mutations are in a particular population) and penetrance (the likelihood of mutation carriers developing cancer). Although thought to be responsible for one half of hereditary breast cancers,2 mutations in these two genes are generally rare. Estimations of mutation prevalence in the U.S population range from approximately 0.1% to 0.76% (1 in 1000 to 1 in 132).7–9 However, in women of Ashkenazi Jewish (Eastern European) descent, approximately 2.5% (1 in 40) are believed to be mutation carriers.10
BRCA1 and BRCA2 mutations have a high penetrance. The lifetime risk of invasive breast cancer with a BRCA1 or BRCA2 mutation ranges from 36% to 85%, depending on the study methodology.11–14 Original data were based primarily on highly selected families such as those used for positional cloning of the genes. In these families, the estimated lifetime risk of breast cancer was over 80%; in BRCA1 carriers, the lifetime risk for ovarian cancer was between 40% and 65%, and for BRCA2 carriers, it was 20%.15,16 Later studies have used casebased ascertainment and, to a lesser extent, population-based data. Population-based designs to ascertain penetrance are usually performed on specific subpopulations (e.g., Ashkenazi Jews) known to harbor a high incidence of founder mutations to obtain an adequate number of carriers. One study of the Ashkenazim showed a 56% lifetime risk (to age 70 years) for breast cancer and a 16% lifetime risk of ovarian cancer but did not distinguish between BRCA1 and BRCA2.10 By comparison, case-based ascertainment usually yields higher penetrance estimates, with 69% and 74% lifetime risk of breast cancer, and 54% and 23% risk of ovarian cancer in Ashkenazi Jews with mutations in BRCA1 and BRCA2, respectively.12 A meta-analysis of case-based studies showed a 65% lifetime risk for breast cancer and 39% risk for ovarian cancer in all-comers with mutations in BRCA1; corresponding values for BRCA2 were 45% and 11%.14 A recent meta-analysis (including both Ashkenazi and non-Ashkenazi Jewish populations) estimated the lifetime risk of breast cancer as 55% in BRCA1 carriers and 47% in BRCA2 carriers; ovarian cancer risk was estimated as 39% in BRCA1 carriers and 17% in BRCA2 carriers.17
Penetrance estimates may also vary, based not only on the populations studied, but also on other risk factors such as oral contraceptive use, parity, and oophorectomy,18 and on the specific location of the mutation within the gene. Analysis of BRCA1 cancer families has revealed a correlation between the mutation site and the relative risk of breast versus ovarian cancer. 3′ Mutations, which cause truncation of the C-terminal region, are associated with a higher proportion of breast than ovarian cancers, whereas 5′ mutations, which delete a large segment of the BRCA1 protein, are associated with a mixture of breast and ovarian cancers.19 Mutations in the central region of BRCA2 (referred to as the “ovarian cancer cluster region”) have been shown to be associated with a decreased risk of breast cancer relative to ovarian cancer risk.20
Features of BRCA1- and BRCA2-Related Cancers
Compared with that of the general population, BRCA1 and BRCA2 mutation carriers have an increased risk of developing breast cancer. The lifetime risk of invasive breast cancer conferred by a BRCA1 or BRCA2 mutation is estimated to be between 36% and 85%. BRCA1- and BRCA2-associated breast cancers are diagnosed at a younger age, often premenopausally.21 This is particularly true for BRCA1 carriers, approximately 20% of whom develop breast cancer before age 40, with 50% by age 50.22 A recent retrospective cohort study indicated that ductal carcinoma in situ (DCIS), with or without invasive cancer, is just as prevalent in mutation carriers (37%) as in high-risk noncarriers (34%) but that it may develop at an earlier age.23 A population-based case-control study looked at the prevalence of BRCA1 and BRCA2 mutations in women diagnosed with DCIS. It was found that mutation prevalence rates in this group were similar to those found in women with invasive breast cancer, suggesting that the criteria used to assess eligibility for screening and risk for positive mutation-carrier status should include diagnoses of DCIS.24 In another cohort study of mutation carriers with stage I or II breast cancer, it was found that the risk of a second primary breast cancer in the contralateral breast was approximately 30% over 10 years, and even higher in women who did not use chemoprevention or undergo oophorectomy.25
In addition to a lower age of onset, BRCA1-related breast cancers typically have features associated with a poorer prognosis, including numerous mitoses and substantial pleomorphism.26 Compared with sporadic and BRCA2-related cancers, BRCA1-type breast cancers characteristically exhibit higher frequency of grade 3 tumors and lower frequency of both estrogen receptors (ER) and progesterone receptors (PR), and they are rarely HER2/neu-positive.27 Approximately 75% of BRCA2-associated breast cancers are hormone receptor–positive, whereas a similar percentage of BRCA1-associated breast cancers are not positive.27 The lack of estrogen, progesterone, and HER2 receptor expression constitutes the “triple-negative” or basal phenotype, which has been identified as having a poorer prognosis than other tumors because of the limited number of therapies that can specifically target these cells.
Compared with the general population, BRCA1 and BRCA2 mutation carriers also have an increased risk of ovarian cancer. The lifetime risk of ovarian cancer in the general population is approximately 1.3%, whereas the lifetime risk of BRCA mutation carriers is estimated to be 10% to 60%. BRCA1 mutations confer a higher risk of ovarian and primary peritoneal cancer compared with risk from BRCA2 mutations, and they are associated with earlier age at onset. Fallopian tube cancers, though much rarer than ovarian cancer, are also higher in carriers than in noncarriers,28 and again this risk is increased in BRCA1 carriers than in BRCA2 carriers. BRCA1– and BRCA2-associated ovarian cancers are pathologically and histologically indistinguishable from one another. However, they are generally of serous histology, with endometrioid being the next most common. Mucinous tumors are unlikely to be associated with BRCA1 or BRCA2 mutations, whereas rarer forms such as clear cell tumors are not common enough for an association (or lack thereof) to be determined to date. In addition, borderline and low-malignant-potential tumors of the ovary are not believed to be associated with BRCA1 or BRCA2 mutations.20
The association of other cancers with BRCA1 and BRCA2 mutations has been studied in a number of cohorts. In particular, BRCA2 mutations are associated with an increased risk of pancreatic cancer and melanoma.10,29 Unfortunately, the lifetime risk of developing these malignancies has not been reliably quantified. A kin-cohort study of unselected patients newly diagnosed with ovarian cancer looked at cancer incidence in first-degree relatives of confirmed BRCA1 and BRCA2 mutation carriers. A higher risk ratio was associated with ovarian, female breast, and testicular cancer in BRCA1 carriers, with higher risk for ovarian, female and male breast, and pancreatic cancers in BRCA2 carriers.8
The precise factors that determine which mutation-positive women will, and will not, develop cancer are unknown. Variation in penetrance of BRCA1 and BRCA2 has resulted in the identification of possible cancer risk modifiers in carriers. Both hormonal and genetic influences have been examined. The use of oral contraceptives, for example, has been reported to protect against ovarian cancer in noncarriers.30 Studies in carriers present differential risk in regard to breast or ovarian cancer, showing a protective effect of oral contraceptives against ovarian cancer31 or no reduction in risk.32 However, a clinical dilemma is presented by data suggesting that the use of oral contraceptives in BRCA1 carriers may also significantly increase breast cancer risk.18 At present, oral contraceptive use is not actively recommended for ovarian cancer prevention, but short-term use for contraceptive needs is not contraindicated.
The relation between endogenous hormonal factors to breast and ovarian cancer risk has also been assessed in BRCA1 and BRCA2 carriers. In a study of the reproductive histories of BRCA1 carriers, the risk of breast cancer was increased in those who experienced menarche before age 12, and in those with parity of less than three.31 It is interesting to note that the risk of ovarian cancer in BRCA1 carriers has been found to be lower with greater parity, in contrast to BRCA2 carriers, in whom parity was associated with a significant increase in ovarian cancer risk.33 Further studies to address these gene-environment and gene-gene interactions are ongoing. These studies are needed to potentially make it possible to provide more personalized risk estimates for the individual woman.
Genetic Testing
Family history features suggestive of a BRCA1 or BRCA2 mutation include premenopausal breast cancer, ovarian cancer, and male breast cancer. A referral for genetic counseling is indicated if the medical or family history is consistent with hereditary breast and ovarian cancer (Box 5-1).
Box 5-1 Features Indicating a Need for Referral for Cancer Genetic Counseling
Breast cancer diagnosed before menopause (typically before age 50)
Member of a family with a known BRCA1 or BRCA2 mutation
Two breast primaries in a single individual, particularly when one was diagnosed premenopausally
Breast and ovarian cancer in a single individual
Personal or family history of male breast cancer
Breast or ovarian cancer in a member of a high-risk population (i.e., Ashkenazim)
Consider the following scenario, which is illustrated in Figure 5-1. Mary is concerned about her risk of developing breast cancer, since her mother (Susan) was recently diagnosed with the disease. Mary’s maternal aunt (Jane) is deceased as a result of breast cancer, suggesting a hereditary component to the family history. If Mary chooses to undergo genetic testing and no mutations are detected (i.e., a negative result), there are two possible explanations for this result. First, Susan’s and Jane’s breast cancers may be due to a mutation in the gene for which Mary was tested. However, because of autosomal dominant inheritance, Mary did not inherit the mutation from Susan. Thus, Mary’s risk of developing breast cancer is likely closer to that of the general population and is based on her own risk factors, since her family history of breast cancer is attributable to a genetic mutation that Mary does not have. However, the second possibility is that Susan’s and Jane’s cancers are due to a mutation in a gene that was not tested or that has not yet been discovered. Therefore, it is not possible to determine whether Mary has the same mutation. In this scenario, Mary remains at an increased risk for breast cancer based on her genetically unexplained family history. It is not possible to distinguish between these two possible explanations without genetically testing Susan.
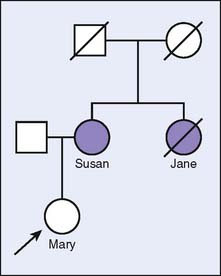
By contrast, if genetic testing were performed on Susan and a mutation were detected, one could reasonably attribute her breast cancer to the identified mutation. Determining whether Mary has this same mutation would then indicate whether she, too, has an increased risk of breast cancer. It can also determine whether Mary’s offspring would have an increased risk of breast cancer.
Genetic analysis of the BRCA1 and BRCA2 genes is clinically available. For most families, full sequencing of both genes is required and is considered the most reliable method of gene analysis. However, an estimated 12% of deleterious mutations are large genomic deletions or duplications, which are not detectable with sequencing.34 Therefore, additional testing technology, such as Southern blot analysis, may be necessary in families with a cancer pattern strongly suggestive of a hereditary component. If a woman with breast cancer undergoes full analysis of both genes and a mutation is identified, the mutation likely explains the most significant genetic component of her cancer. It also indicates that she has an increased risk of ovarian cancer, and intensive screening or consideration of ovarian cancer risk-reducing options is indicated. In addition, it is possible to offer predictive genetic testing to other interested family members to identify those who also have an increased risk of developing breast and ovarian cancer.
In persons of Ashkenazi Jewish descent, genetic testing usually begins with analysis of three founder mutations. The 187delAG and 5385insC mutations in the BRCA1 gene and the 6174delT mutation in the BRCA2 gene account for approximately 90% of BRCA1 and BRCA2 mutations detected in the Ashkenazim. Because of the high detection rate with this threemutation panel, full sequencing is generally considered only in Ashkenazi Jewish persons with a high pre-test probability of a deleterious mutation and whose test results are negative for the three founder mutations. Other populations known to have founder mutations include Icelanders (999del5 in BRCA2) and those from Finland, France, Russia, Denmark, Sweden, Belgium, and the Netherlands.35
Management: Screening and Risk Reduction Options
Breast Cancer
Screening and risk reduction strategies for the breast in female BRCA1 and BRCA2 mutation carriers fall into three general categories: intensive cancer screening, chemoprevention, and prophylactic surgery. According to recommendations of the National Comprehensive Cancer Network (NCCN), women with BRCA1 or BRCA2 mutations should have a clinical breast examination by a healthcare provider at least every 6 months, beginning at age 25 years (www.nccn.org). Mammograms generally begin at age 25 years, but can be adjusted based on the cancer pattern in the family, since there is also the issue of radiation risk. Adjuvant ultrasound is also a consideration for women with dense breast tissue; studies are ongoing to determine its effectiveness.
Recently, magnetic resonance imaging (MRI) has become a more routine part of breast screening for mutation carriers. The American Cancer Society recommends that women with a high risk of developing breast cancer (greater than 20% lifetime risk) should get an MRI and a mammogram every year. The optimal interval for MRIs has not yet been established.36 A review of the effectiveness of MRI as an addition to mammography and ultrasound in screening high-risk young women found consistent evidence that MRI as a screening strategy provides high sensitivity compared with that of mammography alone or mammography and ultrasound with or without clinical breast exam.37 Whether this higher sensitivity translates into a reduction in patient mortality compared with that with mammography alone is presently unclear. This is an evolving field.
Risk reduction options include chemoprevention or prophylactic bilateral mastectomy. Data on the effectiveness of chemoprevention with tamoxifen in BRCA1 and BRCA2 carriers have been extrapolated from large trials within the general population. The National Surgical Adjuvant Breast and Bowel Project (NSABP) prevention trials showed a 62% decrease in the risk of breast cancer in BRCA2-positive women who received tamoxifen for 5 years versus no reduction in breast cancer incidence among BRCA1-positive women.38 However, another study examined the effects of tamoxifen on prevention of contralateral breast cancer in mutation carriers and found that the drug did provide protection overall, but the protection reached significance only in BRCA1 mutation carriers.39 This is an interesting finding in light of the lack of hormone receptor expression seen in BRCA1 relative to BRCA2 cancers and is yet to be replicated.
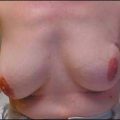
Another risk-reduction option is prophylactic bilateral mastectomy, which reduces the risk of breast cancer by at least 90% in unaffected mutation carriers.21,40 For women with breast cancer who opt for contralateral prophylactic mastectomy, the rate of a contralateral malignancy is also reduced by 90%.41 However, therapeutic and/or contralateral mastectomy does not reduce the risk of chest wall or distant recurrence. Furthermore, although such surgery results in the greatest reduction in breast cancer risk, it is often an emotionally difficult choice for women. Although expected to decrease mortality, the efficacy of bilateral prophylactic mastectomy in prolonging survival has not yet been shown in prospective studies.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


