Keywords
T cell receptor, chimeric antigen receptor, cancer, adoptive immunotherapy, gene therapy
Introduction
There are two types of immune responses: humoral immunity and cellular immunity. Humoral immunity is mediated by antibodies generated by B cells. Antibodies have high binding affinity to specific antigens and usually bind to three-dimensional epitopes that are expressed on the surface of target cells. Cellular immunity requires cell-to-cell contact for recognition and destruction of target cells. Cellular immunity is mediated by T lymphocytes, natural killer (NK) cells, and other cells of the immune system. T cells have antigen specificity that is mediated by the T cell receptor (TCR), a membrane heterodimer that is associated with the CD3 signaling complex. The TCR binds peptides, processed from pathogens and malignant cells, which are bound to proteins encoded by genes in the major histocompatibility complex (MHC). Other components of the cellular immune response, such as NK cells, recognize targets via receptors whose ligands can be expressed on many normal, virus-infected, and malignant cells.
One key hurdle to overcome for effective immunity against virus-infected and tumor cells is eliciting a strong immune response. Most viruses and tumors have developed mechanisms to evade the host immune response. Therefore, patients with chronic virus infections and malignancies often have a weak and ineffective immune response to their viruses or tumor cells.
It has been known for some time that passive transfer of humoral immunity, in the form of γ-globulin injections, can boost the immune response to pathogens. During approximately the past decade, it has become clear that the transfer of passive immunity to patients can be effective in treating malignancies. Herceptin, Avastin, and others represent the passive transfer of humoral immunity. Adoptive transfer of lymphokine-activated lymphocytes, tumor-infiltrating lymphocytes (TILs), and ex vivo expanded antigen-specific PBL-derived T cells can mediate objective clinical responses, especially when infused into patients who were pretreated with nonmyeloablative chemotherapy. However, it is difficult to isolate tumor-reactive T cells from most cancer patients, and it takes time to expand these cells to therapeutic numbers ex vivo . Therefore, adoptive cell transfer has limited potential for treating cancer patients.
The difficulty in raising tumor-reactive T cells, combined with the time it takes to expand these cells to therapeutic numbers, has led many in the field to consider the use of genetically modified T cells as an alternative approach for treating cancer patients. Chimeric antigens receptors (CARs) and TCRs are most commonly genes used to redirect the specificity of T cells. However, there are many aspects of CAR and TCR engineered T cells that must be resolved before these effectors become a standard treatment for cancer patients.
Chimeric Antigen Receptors
The first group of antigen receptors that have been widely studied for use in the genetic modification of T cells were CARs. A CAR combines an extracellular binding domain that recognizes and binds a specific antigen with an intracellular domain that transfers cellular signals . The antigen recognition domains are derived from the antigen binding fragment (Fab) of mouse monoclonal antibodies (mAbs) that have high affinity for the specific antigens ( Figure 18.1 ). In a mAb, the Fab fragment consists of the variable domains of both heavy chain and light chain. The Fab fragment in a CAR is generated as a single-chain variable fragment (scFv) via a short linker fragment that binds heavy- and light-chain variable domains . The intracellular domain of CARs contains the signaling domain of the zeta chain of the TCR/CD3 complex or the gamma chain of the immunoglobulin receptor FcεRI , which transduces cellular signals by phosphorylating their immunoreceptor tyrosine-based activation motif (ITAM) ( Figure 18.1 ). A hinge region that connects the antigen binding and signaling domains of a CAR is also included to provide some degree of flexibility and a distance between the epitope and the target cell membrane for optimal T cell activation .
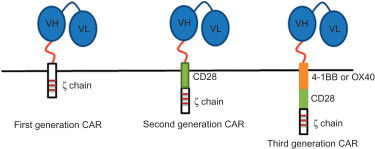
There are distinct advantages to using CAR-transduced T cells for adoptive immunotherapy. Because CARs bind to antigen in the same way as their parent mAb, CAR-mediated signaling is not dependent on the expression of MHC molecules or antigen processing and presentation . CARs also contain all of the elements needed to bind antigen and activate the T cell, so they do not need to associate with the CD3 complex for T cell activation and function. Being a single-chain construct, CAR constructs are compact and the vectors are relatively small, making it easy to make high-titer virus. Also, single-chain CARs are not subject to mispairing with other chains expressed by the T cells.
There are some limitations to the use of CAR-engineered T cells . For example, these cells can only recognize antigens on the surface of target cells. Also, the binding of antibody to antigen is much stronger than the binding of TCR and antigens, which may impact how the activation signals influence T cell function . In addition, being chimeric molecules, there is potential immunogenicity for these chimeric receptors . In this section, we discuss factors that influence CAR expression and function as well as highlight the results of key animal and human studies.
Targets
One of the attractive features of CARs is that their antibody specificity allows them to recognize structures on intact proteins, carbohydrates, and lipids. As a result, the target antigens do not need to be processed and presented by MHC molecules, and any molecule on the cell surface can be easily targeted by CARs. The list of CAR targets evaluated includes but is not limited to CD19 , CD20 , and others for hematologic malignancies; Her-2 , MUC-1 , CEA , PSMA , and others for breast cancer, colon cancer, prostate cancer, and other solid tumors; and VEGF-R2 and KDR for tumor neovasculature. Essentially, any mAb can be engineered to make a CAR. For those targets where it is difficult to raise conventional antibodies in mice or other species, phage display and other technologies have been used to generate the antigen binding portion of the CAR . Therefore, CAR-based therapeutic options are limited only by the availability of mAbs.
Design
The general CAR design described previously consists of an scFv fragment fused to the hinge region of CD8, which is fused to the intracellular signaling regions from Fcγ or CD3ζ molecules ( Figure 18.1 ). Any CAR with this basic design is considered to be a “first-generation” CAR. Although first-generation CARs effectively transfer antitumor reactivity to mouse and human T cells, they have limited proliferative potential and persistence in vivo . A potential explanation for this is that CARs bypass all of the normal costimulatory signals that T cells use for survival and memory formation. One solution to these problems was the inclusion of a costimulatory cassettes (e.g., CD28, 4-1BB, OX40, or ICOS) in the CAR construct ( Figure 18.1 ). By having two signaling cassettes, one T cell activating and one costimulatory, T cells engineered with second-generation CAR constructs have enhanced proliferative capacity and persistence in vivo compared to T cells engineered with first-generation CA’s . Recently, a third-generation CAR has been developed that includes a third signaling cassette. In addition to the CD3ζ signaling cassette, two costimulation cassettes (CD28, 4-1BB, OX40, and/or ICOS) are included . T cells engineered with third-generation CARs have superior function, proliferative capacity, and survival in vivo compared to T cells engineered with second-generation CARs .
Another consideration in CAR design is the relative affinity of the scFV. In general, Fab fragments have higher affinity for antigen than TCRs have for peptide MHC complexes . However, the use of phase display libraries has led to high-affinity mAbs for CAR generation . These high-affinity CARs can bind to very low levels of antigen, making the CAR gene-modified T cells extremely potent antitumor effectors.
In Vivo Studies
T cells engineered with CARs have been extensively evaluated in vivo . In mice, it is clear that CAR gene-modified T cells can treat established tumors. Mouse T cells transduced to express first-generation CARs can reject established mouse tumors and human xenografts in vivo . In addition to using CAR-engineered T cells, several studies report using CARs to engineer hematopoietic stem cells . Mice that were engrafted with CAR-engineered hematopoietic stem cells were resistant to subsequent tumor challenge . One unexpected observation in these experiments was that a CAR can be expressed and function in non-T cell subsets such as cells of the myeloid lineage and NK cells . These studies indicate that CARs are effective therapeutic agents and have the added advantage of functioning in multiple effector lineages.
Although promising results were obtained with first-generation CAR gene-modified T cells, there were clear limitation in persistence noted in vivo . In cancer patients, CD3ζ chain downregulation and modulation of CD8 coreceptor chains have been implicated in T cell dysfunction. The advantage of using CARs to redirect the specificity of T cells is that the resulting CAR-expressing T cells are not dependent on many of the signaling molecules (CD3, CD4, or CD8) that are required for TCR signaling. However, the disadvantage is that CAR-expressing T cells may not get the necessary costimulatory signals needed for T cell survival in vivo . The second-generation CAR, which includes a CD28 signaling cassette, solves this problem ( Figure 18.1 ). TNP CAR transgenic mice expressing a CD28 cassette or a truncated CD28 cassette lacking the CD28 intracellular signaling domain demonstrated that costimulation was essential for full activation of CAR-expressing T and NK cells . Moreover, CD28 costimulation led to improved effector function and resistance to apoptosis of CAR-expressing T cells . Direct comparisons of first- and second-generation CAR-engineered T cells found that second-generation CAR-expressing T cells persisted longer in vivo and had better antitumor activity than first-generation CAR-expressing T cells . Recent studies with third-generation CARs indicate that multiple costimulation signaling domains are an improvement over one (second generation) or none (first generation) . The combination of CD137 (4-1BB) and CD28 in these third-generation CARs led to enhanced persistence and rejection of large tumors . Collectively, these studies indicate that the addition of costimulation leads to improved function and therapeutic efficacy of CAR-expressing T cells in animal models.
Several different CARs have been used to genetically modify T cells for use in clinical trials in patients with cancer and infectious diseases. The first clinical trials used a first-generation CAR targeting carbonic anhydrase IX (CAIX) for patients with renal cell carcinoma and folate binding protein (FBP) for patients with ovarian cancer . These studies demonstrated that it is feasible to prepare CAR-engineered autologous T cells for patient treatment. In both studies, no objective clinical responses were observed, and the gene-modified T cells failed to persist long term. Although no serious adverse events were observed using the FBP CAR gene-modified T cells, there was grade 2–4 liver toxicity in the patients treated with CAIX CAR gene-modified T cells. This toxicity was due to the expression of the CAIX antigen on the epithelial cells of the bile duct . In a later trial, a patient was treated with a third-generation CAR (ssFv/CD28/4-1BB/CD3ζ) targeting ERB B2 (Her-2) . The patient rapidly developed respiratory distress and died 5 days later. It was found that there was low-level ERB B2 expression on the epithelial cell of the lung, and the toxicity was presumably due to destruction of the normal lung epithelium . These results indicated that selection of the target and probably the affinity of the CAR are critical considerations for pursuing clinical trials using CAR gene-modified T cells.
Whereas there have been adverse events associated with CAR gene-modified T cells, there have been many encouraging clinical trials reported. Clinical trials have been reported using T cells expressing CARs targeting CD19, CD20, GD2 (Brenner), and others . The best characterized of all of these CARs targets CD19 on B cell lymphomas. Several groups have conducted clinical trials with gene-modified T cells and have obtained very promising results . In all cases, the only normal cells found to express CD19 are normal B cells, and the CD19 CAR can impact on number of circulating B cells in patients after treatment, leading to B cell aplasia . Other side effects reported in a few patients included tumor lysis syndrome and cytokine storm . The conclusions from these clinical trials indicate that T cells expressing anti-CD19 CAR were well tolerated and resulted in few adverse events.
T Cell Receptors
A second group of antigen receptors used to engineer T cells is the TCRs. CD8 + and CD4 + T cells express a TCR αβ heterodimer that recognizes antigenic peptide fragments bound to MHC class I or class II, respectively. TCR genes were first used to redirect T cell specificity in mice . The expression of transduced α and β genes of TCRs confers upon the recipient cells the specificity of the donor T cells. Although TCR genes have been cloned and expressed in hybridomas for years, engineering human T cells with TCR genes is a relatively recent occurrence. We first demonstrated that normal donor PBL-derived human T cells could be engineered to recognize HLA-A2 + MART-1 + melanoma cells . Since then, many critical advances have been made in TCR gene transfer technology. Being heterodimers, TCRs differ from CARs in that two chains need to be expressed rather than just one to redirect the specificity of a T cell. There are also issues related to the competition with the endogenous TCRs for expression and pairing. Finally, TCRs are restricted by a single MHC molecule, restricting the number of patients who can be treated using any single TCR. In this section, we discuss how challenges with TCR expression and function have been overcome and the early results from clinical trials.
Targets
Since we first reported engineering normal human PBL-derived T cells with TCR genes, the number of TCR genes evaluated has grown to include TCRs that target other melanoma differentiation antigens (gp100 and tyrosinase ), cancer testis antigens (NY-ESO-I and MAGE ), viral antigens (CMV , EBV , HIV , HCV , and others), and other tumor antigens (CEA , p53 , HA-1/HA-2 , and others). As with CARs, the potential targets of TCR gene-modified T cells are limited only by the availability of antigen-reactive T cell clones and TCR genes. Unlike isolating antibodies for generating CARs, raising T cell clones against many target antigens on tumors has been difficult. This problem can be circumvented by using T cell clones isolated in HLA-A2 transgenic mice that recognize human tumors or by using phage display to generate single-chain TCRs . Like CARs, the spectrum of TCR gene-modified T cells is limited only by the availability of TCR genes.
Factors Influencing TCR Assembly and Function
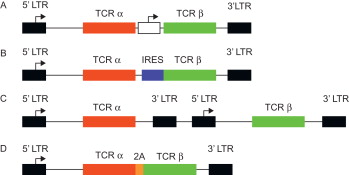
The proper function of a TCR gene-modified T cell requires adequate expression of the introduced TCR α and β chains, correct pairing of the TCR chains, and proper pairing with the elements of the CD3 complex. Initially, vectors containing multiple promoters ( Figure 18.2A ) , IRES elements ( Figure 18.2B ) , or multiple vectors ( Figure 18.2C ) were used to ensure adequate expression of both TCR chains. Recently, the use of 2A self-cleavage peptides has ensured that both chains are equally expressed from a single promoter ( Figure 18.2D ) . As shown in Figure 18.3 , 2A linked TCR chains generally give higher and more uniform TCR expression compared to having the chains expressed separately using individual promoters. It has also been shown that high TCR transgene expression can be achieved by codon optimizing the TCR α and β chain genes . The use of 2A sequences and codon optimization also has the advantage of increasing TCR expression without altering the TCR sequence.
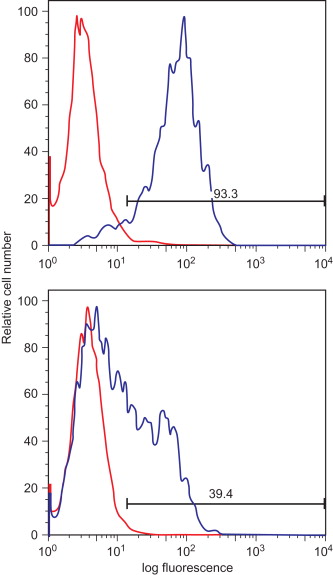
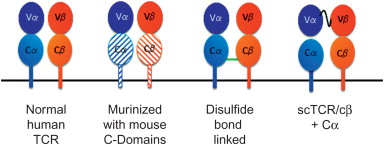
High and equal expression of the TCR α and β chains is not adequate to ensure a functional TCR is expressed on the surface of T cells. TCR expression is limited by the components of CD3 complex. Because each mature T cell has its own TCR, there is competition for the CD3 complex in the T cell surface, thus limiting TCR transgene expression. Also, pairing between the endogenous and exogenous TCR chains further limits the amount of introduced TCR expressed. The issue of TCR assembly has been addressed in several ways. We noticed that human TCR genes have better expression and function when their human constant regions are substituted with mouse constant regions . It was determined that inclusion of elements of the mouse TCR constant regions led to more efficiently pairing between the TCR chains and with the CD3 complex . Altering the glycosylation of the TCR , adding cysteine residues to the constant regions to promote interchain disulfide bonds , and inclusion of leucine zippers have been shown to facilitate proper pairing of the introduced TCR chains, leading to enhanced TCR expression and function.
Another critical factor that influences a TCR’s ability to redirect the specificity of T cells is affinity. During our initial studies, we found that our HLA-A2-restricted, MART-1-reactive TCR could engineer normal CD8 + T cells to recognize peptide-loaded targets and melanoma cells, whereas TCR-transduced CD4 + T cells could recognize peptide-loaded targets but not melanoma cells . This dependence on the CD8 coreceptor for tumor cell recognition led us to postulate that if a TCR could be identified with high affinity, we could generate “better” CD8 + T cells and MHC class I-restricted CD4 + T cells . We and subsequently others were successful in identifying CD8-independent TCRs that could transfer MHC class I reactivity to CD4 + T cells . These high-affinity TCRs have the added benefit of requiring a lower level of expression to activate T cells, circumventing some of the expression issues described previously.
Naturally occurring CD8-independent TCRs are relatively rare, so they are difficult to find. It has been shown that some of the previously described modifications designed to enhance expression and function can also convert a CD8-dependent TCR to a CD8-independent TCR . Also, tumor-reactive T cell clones in transgenic mice expressing human HLA-A2 molecules can express CD8-independent TCRs . However, the field has generally taken the approach of modifying the CDR 3 regions of existing TCRs to convert them to high affinity and CD8 independence . High-affinity TCRs continue to be identified and evaluated for their use in TCR gene transfer studies.
There are clear advantages to using CD8-independent/high-affinity TCRs. First, lower TCR transgene expression is needed to activate the TCR gene-modified T cells. Second, CD8-independent/high-affinity TCRs are able to generate a novel population of MHC class I-restricted CD4 + T cells . However, the question of need for CD8-independent/high-affinity TCRs for engineering T cells has not been resolved. We investigated the impact on TCR affinity and tumor cell recognition by T cells . We found that by increasing the stability of a low-affinity TCR/peptide/MHC trimolecular complex, which essentially mimics increasing the affinity of the TCR, we got better antitumor reactivity. However, increasing the stability of a high-affinity TCR/peptide/MHC trimolecular complex did not improve T cell reactivity, suggesting there is an upper limit to which TCR affinity impacts T cell function. We argued that improved signaling in the T cell led to better function in T cells expressing high-affinity TCRs . The importance of T cell biology rather than TCR affinity makes sense given the TIL studies and the impact of immune suppression on T cells. The affinity of the TCRs is no different on TIL cells in the tumor lesion versus TIL cells infused into patients following ex vivo expansion, but their impact on disease can be quite different. Similarly, T cells bearing high-affinity receptors can be rendered inactive in the presence of Treg cells or myeloid-derived suppressor cells. Despite these arguments, it still makes sense to use high-affinity TCRs for TCR gene transfer studies if they are available.
In Vivo Studies
Adoptive transfer of TCR gene-modified T cells has been evaluated in mice. Infusion of TCR-transduced splenocytes led to rejection of tumors and decreased viral load, indicating that the TCR gene transfer approach could be effective in vivo . In addition, there is a report of engineering Treg cells to suppress unwanted immune responses . Most reports found no adverse events in mice , but recently there have been indications that TCR gene-modified T cells could lead to adverse events, presumably due to TCR mispairing . Although there remains debate in the field regarding the impact of TCR mispairing on adverse events, there are solutions to the potential problem of off-target reactivity of TCR-transduced T cells. Anything that facilitates pairing of the introduced TCR chains, such as introducing disulfide bridges ( Figure 18.4 ) , leucine zippers , or mouse constant regions ( Figure 18.4 ) into human TCRs , would limit mixed pairing. Two other clever approaches have been described that limit mixed pairing. It has been shown that when TCR αβ chains are introduced into γδ T cells, there is no mixed chain pairing, leading to effector cells with less chance of promoting off-target toxicity . Another approach to limit mixed chain pairing involved altering the structure of the introduced TCR chains . As shown in Figure 18.4 , a normal TCR consists of an α and a β chain, with each chain containing one variable region and one constant region. By fusing the Vα region to the Vβ and Cβ regions, a three-domain chain is generated ( Figure 18.4 ). In order for this construct to express on the surface of T cells, a Cα single-domain chain must be coexpressed. Because neither of these chains can pair with the endogenous TCR and only with each other, this would be an effective approach for eliminating adverse events due to chain mispairing.
TCR gene-modified T cells have also been evaluated in humans. The first two clinical trials reporting the use of TCR gene-modified T cells to treat cancer patients were published in 2006 . The first trial used T cells engineered with the MART-1-reactive TCRs we first described . Allogeneic anti-MART-1 TCR-transduced T cells were injected into melanoma tumor lesions . The goal of this study was to destroy the treated lesion and induce systemic anti-melanoma immunity through cross priming. The second trial used T cells engineered with a different MART-1 TCR for systemic infusion into lymphodepleted stage IV melanoma patients . Both of these trials had objective clinical responses (1 of 15 and 2 of 17 patients, respectively) and indicated that TCR gene-modified T cells were generally safe and well tolerated by the patients.
There have been several additional reports of clinical trials using TCR gene-modified T cells in cancer patients. One report targeted the melanoma differentiation antigens MART-1 and gp100 . The anti-MART-1 TCR used in this trial had higher affinity than the two used in the previous trials, and there was an increase in the frequency of clinical responses (6 of 20 patients treated) . The anti-gp100 TCR reported in this study was of mouse origin and was selected for CD8 coreceptor-independent tumor cell recognition . Of the 16 patients treated with gp100 TCR-transduced T cells, 3 had objective clinical responses. Although no off-target adverse events were reported, the use of T cells engineered with these high-affinity TCRs was implicated with the observed toxicity that was associated with the destruction of normal melanocytes . Many patients had melanocyte destruction in the skin, eye, and ear, resulting in vitiligo, uveitis, and hearing impairment, respectively. Vitiligo (skin depigmentation due to melanocyte destruction) is not reversible with any known treatment and is considered a common side effect in patients treated with interleukin-2 (IL-2) and other forms of immunotherapy . Vision and hearing changes were unexpected side effects of the therapy that were resolved in almost all patients naturally or with use of topical steroids . Although high-affinity TCRs did lead to improved clinical outcomes for melanoma patients, the adverse events need to be closely monitored in similar studies in the future.
A major advantage of the TCR gene-modified T cells for treating cancer patients is that it is possible to treat any patient with autologous tumor-reactive T cell regardless of their natural capacity to develop an antitumor immune response. Raising CEA-reactive T cells with antitumor reactivity for patient treatment has proven to be challenging. A clinical trial was reported using a mouse anti-CEA TCR to engineer human T cells to treat patients with advanced colon carcinoma . All three patients treated had a transient decrease in serum CEA levels, and one patient had a partial clinical response. However, all patients had severe inflammatory colitis with grade 2 (one patient) and grade 3 (two patients) diarrhea. Despite the interesting clinical results, due to toxicity associated with this trial, enrollment of additional patients was halted.
The adverse events associated with some TCR gene transfer trials led to testing targets considered to be “tumor specific.” One attractive group of targets is the cancer testis antigens . TCRs have been isolated that target NY-ESO-I, MAGE-A3, MAGE-A4, and other cancer testis antigens . T cells engineered to express an NY-ESO-I TCR have been used to treat patients with melanoma and synovial cell sarcoma . Objective clinical responses were reported in 5 of the 11 melanoma patients and 4 of the 6 synovial cell sarcoma patients treated with NY-ESO-I TCR-transduced autologous T cells . None of the patients exhibited any of the adverse events (skin, eye, and hearing) reported in the previous trials. Therefore, it is clear that depending on the antigen targeted by TCR gene-modified T cells, the TCR gene transfer approach can be a safe and potentially effective treatment for advanced malignancies.
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Clinical Application of Plasmid-Based Cancer Vaccines
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Clinical Application of Plasmid-Based Cancer Vaccines
 Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
Imaging of Oncolytic Virus Gene Expression
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


