GENETIC SUSCEPTIBILITY TO RENAL CELL CARCINOMA
von Hippel-Lindau Disease
VHL disease is an autosomal dominant condition that affects approximately 1 in 36,000 live births worldwide. The VHL gene is located on the short arm of chromosome 3 (3p25) and is the only known susceptibility locus associated with the condition. It is a well-studied tumor suppressor gene that demonstrates loss of heterozygosity in RCCs of patients with VHL disease and sporadic clear cell RCC as well.
VHL disease is a multisystem condition, and an affected individual is at risk to develop any of the following lesions: (1) hemangioblastoma of the cerebellum, spine, or retina; (2) papillary cystadenoma of the epididymis, the adnexal organs, or the endolymphatic sac; (3) adrenal pheochromocytoma and occasionally extra-adrenal paraganglioma; (4) pancreatic cysts, serous cystadenomas, and neuroendocrine tumors (NET); and (5) multiple and/or bilateral RCC and cysts.
Although the penetrance of VHL disease is 100%, where individuals will develop at least one associated lesion by their sixth decade of life, the expressivity is highly variable even among individuals sharing the same gene mutation. The disease is phenotypically categorized into type 1 and type 2 based on risk for developing pheochromocytoma, with the latter further divided into three subtypes (2A, 2B, and 2C) based on risk for developing RCC. The genotype/phenotype correlations within each type are described in Table 33.2.
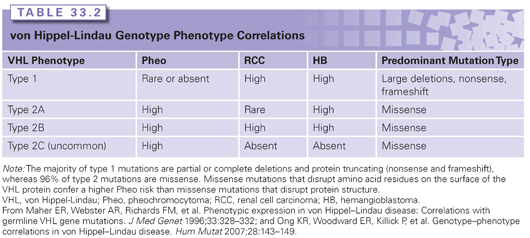
Renal Lesions
RCCs of patients with VHL disease are of exclusively clear cell histology. The lifetime risk for developing RCC is 25% to 45%, and when renal cysts are included, the risk rises to 60%.5 Renal cysts and RCCs develop at an earlier age in patients with VHL in comparison to sporadic counterparts, with an average age of 39 years (range, 16 to 67 years).5 Cystic lesions are typically asymptomatic; however, complex cysts must be monitored closely with computed tomography (CT) or magnetic resonance imaging as they will harbor a visibly solid RCC component. RCC will often arise from noncystic parenchyma as well.
Nonrenal Clinical Features
With the exception of RCC and pancreatic NETs, the malignancy risk with VHL-associated tumors is very low. Renal lesions and hemangioblastoma of the cerebellum, spine, or retina are common presenting lesions in VHL. The risk for developing a single hemangioblastoma of the spine, cerebellum, and brainstem is 60% to 80%, and the average age is 33 years (range, 9 to 73 years),5 although most patients can develop multiple lesions at any point in their lifetime. Patients may remain completely asymptomatic, especially during periods of no growth or slow growth. Surgical resection is delayed until onset of symptoms.
Retinal hemangioblastomas (retinal angiomas) are usually multifocal and bilateral. These hypervascular tumors can lead to retinal detachment and vision loss. Retinal hemangioblastomas have been observed in 25% to 60% of patients with an average age of 25 years (range, 1 to 67 years). Approximately 5% of lesions are seen younger than 10 years, making genetic testing of at-risk children essential as affected children should undergo annual retinal examinations beginning at birth. Pheochromocytoma has also been observed in young children and can present as a hypertensive crisis. The average age at presentation is 30 years (range, 5 to 58 years), and the risk is 10% to 20%.5
Pancreatic manifestations include multiple simple cysts and serous cystadenomas (47% and 11%, respectively), which follow a benign course and are almost always asymptomatic in patients. Pancreatic NETs are less common (15%); however, approximately 2% undergo malignant transformation.6 A NET tends to be indolent and is seldom the initial presenting lesion; however, close monitoring is indicated for timing of surgical resection. A less common manifestation of VHL is a papillary cystadenoma of the endolymphatic sac, or inner ear, which is extremely rare in the general population but more prevalent in VHL disease (~11%). Papillary cystadenomas may also arise in the epididymis in men and less commonly in the adnexal organs in women.
von Hippel-Lindau Molecular Genetics
The VHL gene was cloned by Latif et al.7 in 1993 and is the most well studied of the familial RCC syndromes. Loss of VHL function has been demonstrated to cause RCC formation in VHL disease as well as in the majority of sporadic clear cell RCCs.8,9 The VHL gene encodes the pVHL protein, which in normoxic conditions forms a complex with elongin B, elongin C, Cullin 2, and Rbx1. The VHL complex targets HIF-1α and HIF-2α for ubiquitin-mediated degradation. The HIF-1α and HIF-2α genes, along with HIF-3α, encode the α subunit of the HIF heterodimer. In hypoxic conditions, the VHL complex does not interact with HIF-1α and HIF-2α, leading to an accumulation of these subunits and downstream transcription of HIF-dependent genes. Loss of VHL protein function in renal tumors simulates low tissue oxygen levels, or “pseudohypoxia,” where HIF-1α and HIF-2α accumulate, causing upregulation of many genes involved in tumorigenesis such as vascular endothelial growth factor (proangiogenesis), epidermal growth factor receptor (cell proliferation and survival), and glucose transporter 1 (regulation of glucose uptake).
von Hippel-Lindau Genetic Testing
Genetic testing of the VHL gene is available on a clinical basis and involves full-gene sequencing and large gene rearrangement analysis. When both methods are used, the mutation detection rate is nearly 100% in patients with a clinical diagnosis of VHL.10 Approximately 80% of patients have a parent with VHL, and ~20% represent de novo cases where neither parent carries the mutation. Genetic testing is recommended for a proband with a personal and family history of VHL, as the identification of causative mutations aids in determining disease subtype (see Table 33.2). Disease subtype information along with a careful, detailed family history aids in guiding screening and surveillance of patients with VHL. In simplex cases, where a patient has two or more VHL-associated lesions and a negative family history, genetic testing is recommended to establish a diagnosis. When a mutation is identified in a proband, at-risk family members should be offered predictive testing. Since young children with VHL are known to be at risk for retinal lesions and pheochromocytoma, genetic testing should be offered any time after birth.
Birt-Hogg-Dubé Syndrome
In 1977, Drs. Birt, Hogg, and Dubé first described a multigenerational kindred showing autosomal dominant transmission of fibrofolliculomas with trichodiscomas and acrochordons.11 The phenotype was later expanded beyond dermatologic manifestations to include lung cysts and pneumothorax and renal tumors.12 The number of families with BHD syndrome described in the literature to date is small, and therefore, the exact incidence is unknown. Inherited mutations in the folliculin (FLCN) gene are associated with BHD syndrome.
Renal Lesions
An individual with BHD syndrome is at increased risk of developing multiple and bilateral renal tumors, frequently of more than one histologic type even within the same renal unit, and at younger ages compared with the general population. The lifetime risk is in the range of 27% to 45%,13,14 and the wide range may be a reflection of ascertainment bias introduced when families are recruited predominantly through dermatology clinics versus urology. The most common tumor pathology found in patients is a hybrid oncocytic RCC, which contains a mixture of oncocytic and chromophobe cells. Furthermore, radical nephrectomy specimens of patients have demonstrated oncocytosis where tiny nodules of cells similar to the larger hybrid tumors are diffusely scattered throughout the renal parenchyma. A retrospective study by Pavlovich et al.15 examined 130 tumor specimens from 30 patients (25 males, 5 females) in 19 different BHD families. The authors found that hybrid oncocytic (50%) and chromophobe (34%) were the more common histologic findings, followed by clear cell (9%), benign oncocytoma (5%), and papillary (2%). The average age at first tumor was 50.7 years, and patients averaged 5.3 tumors each (range, 1 to 28). Other studies reporting histologic subtypes of BHD syndrome–related renal tumors have similar findings.
Nonrenal Manifestations
Skin findings associated with BHD syndrome are benign and consist of fibrofolliculoma, trichodiscoma (which are histologically and clinically indistinguishable from angiofibroma), perifollicular fibroma, and acrochordons. Fibrofolliculoma is highly specific for BHD syndrome, whereas trichodiscomas and acrochordons are not. Onset for skin lesions is typically at older than 25 years, and a dermatologic diagnosis of BHD syndrome can be made on the basis of the presence of five or more facial or truncal papules with at least one histologically confirmed fibrofolliculoma.
Approximately 83% to 89% of patients with BHD syndrome will have multiple pulmonary cysts14,16,17 identified upon chest CT. The lifetime risk of spontaneous pneumothorax is 24% to 32%,14,18 and the majority of patients have their first event by age 50 years. The presence of lung cysts is strongly associated with risk of pneumothorax,17 but the mechanism behind this is not known. A possible association between BHD syndrome and parotid oncocytoma has also been reported in a small number of cases.14
FLCN Molecular Genetics
The FLCN gene is located on chromosome 17p11.2 and was cloned by Nickerson et al.12 in 2002. The gene has 14 exons and encodes the protein folliculin. The role of FLCN and tumorigenesis has not been fully established, but animal studies and loss of heterozygosity studies in renal tumors provide some evidence that it is a tumor suppressor gene. Folliculin binds with folliculin-interacting proteins (FNIP1 and FNIP2) and then binds AMP-activated protein kinase, which is part of the cellular energy and nutrient sensing system. AMP-activated protein kinase also helps regulates mTOR activity (mTORC1 and mTORC2). Studies of renal tumors from heterozygous BHD knockout mice and renal tumors from patients with BHD syndrome show mTOR activation. Therapeutic agents inhibiting mTOR activity in sporadic chromophobe tumors are currently under investigation and may have implications for patients with BHD syndrome–related renal tumors.19
The mutation detection rate for FLCN clinical testing is approximately 89%, and nearly all of the mutations described to date have been truncating point mutations (frameshift and nonsense). Splice-site mutations have also been reported in a small number of BHD families, and one missense mutation in a patient with bilateral renal tumors has been reported as well.20 A mutational hotspot in a polycytosine tract in exon 11 has been suggested.14
Hereditary Papillary Renal Cell Carcinoma
HPRCC is inherited in an autosomal dominant manner with reduced penetrance where patients with HPRCC are at risk of developing multiple and/or bilateral papillary RCCs at a young age. The phenotype is limited to the risk of papillary RCC alone, particularly papillary type 1, although the distinction between type 1 and type 2 is not always made on initial pathology review.
Germ-line mutations in the c-met or MET proto-oncogene on chromosome 7q31.2 have been associated with HPRCC.21
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


