MULTIPLE ENDOCRINE NEOPLASIA TYPE 1
Syndrome Description
Multiple endocrine neoplasia (MEN) type 1 is an autosomal dominant syndrome with an estimated incidence in the general population on the order of 1/100,000 to 10/100,000.1,2 The major endocrine features of MEN1 are parathyroid adenomas, enteropancreatic endocrine tumors, and pituitary tumors. A diagnosis of MEN1 is made in a person with two of the three major endocrine tumors, or in an individual with at least one of these tumors if another relative has a diagnosis of MEN1.3–5 The age-related penetrance of MEN1 is 45% at the age of 30 years, 82% at the age of 50 years, and 96% at the age of 70 years.5–7
The most common feature of MEN1 is parathyroid adenoma, which results in primary hyperparathyroidism (PHP). In approximately 50% to 85% of patients with MEN1, PHP will be the presenting manifestation. These tumors occur in 80% to 95% of patients by the age of 50 years,5,8–10 and are typically multiglandular and often hyperplastic.1 The average age at onset of PHP in MEN1 is 20 to 25 years, in contrast to that in the general population, which is in the 50s. Parathyroid carcinoma in MEN1 is rare but has been described.11–13
Pancreatic endocrine tumors are the second most common endocrine manifestation in MEN1, occurring in up to 30% to 80% of patients.5,8 Gastrinomas and insulinomas are most common, followed by VIPomas (vasoactive intestinal peptide), glucagonomas, and somatostatinomas. These tumors are usually multicentric and can arise in the pancreas or more commonly as small (<0.5 cm) foci throughout the duodenum.14 Gastrinomas represent 50% of the gastrointestinal neuroendocrine tumors in MEN1 and are the major cause of morbidity and mortality in patients with MEN1.5,15 Most result in peptic ulcer disease (Zollinger–Ellison syndrome), and half are malignant at the time of diagnosis.14–16 Nonfunctional tumors of the enteropancreas, some of which produce pancreatic polypeptide, are seen in 20% of patients.17–19
Approximately 15% to 50% of patients with MEN1 will develop a pituitary tumor.5,8 Two-thirds are microadenomas (<1 cm in diameter), and the majority are prolactin secreting.20 Other manifestations of MEN1 include carcinoids of the foregut (typically bronchial or thymic), skin lipomas, facial angiomas, and collagenomas and adrenal cortical lesions, including cortical adenomas, diffuse or nodular hyperplasia, or rarely carcinoma.7,21 Thyroid adenomas, pheochromocytoma (PC) (usually unilateral), spinal ependymoma, and leiomyoma have also been reported.22
Genetic Testing
MEN1 is caused by mutations in the MEN1 gene, which is located on chromosome 11q13. It is inherited in an autosomal dominant manner. Germline mutations are typically found in 80% to 95% of families with two or more affected members and in up to 65% of simplex cases (single case of MEN1 with no family history).4,23 Menin, the protein encoded by the MEN1 gene, functions as a tumor suppressor gene and is involved in multiple cellular functions including transcription regulation, genomic stability, cell division, and cell cycle control.24
More than 1,100 mutations have been identified in the MEN1 gene to date, and these are scattered across the entire coding region.24 The majority of these are nonsense or frameshift mutations, and the remainder are missense or in-frame deletions that lead to expression of an altered protein. Splice-site mutations have also been described. There is currently no evidence of genotype–phenotype correlations, and interfamilial and intrafamilial variability is the rule.25,26
Whereas the MEN1 mutation detection rate is quite high in simplex and familial cases, the greater diagnostic challenge for the clinician is when to order genetic testing in an individual who does not meet diagnostic criteria, but has one of the three component tumors. The prevalence of MEN1 among patients with apparently sporadic component tumors varies widely by tumor type. Approximately one-third of patients with Zollinger–Ellison syndrome will carry an MEN1 mutation.27,28 In individuals with apparently isolated hyperparathyroidism (HPT) or pituitary adenomas, the mutation prevalence is lower, on the order of 2% to 5% for each,20,29,30 but the prevalence is higher in individuals diagnosed with these tumors at younger ages (<30 years old). In a small series of patients with isolated foregut/midgut carcinoids, none of 68 were found to carry an MEN1 mutation. Some authors suggest MEN1 testing in those not meeting diagnostic criteria if one of the following is present: Gastrinoma at any age, multifocal pancreatic islet cell tumors at any age, parathyroid adenomas before the age of 30 years, multiglandular parathyroid adenomas, or recurrent HPT, or in individuals with one of the three main MEN1 tumors plus one of the less common tumors/findings.31
Management
Screening and surveillance for MEN1 should use a combination of biochemical screening and imaging as follows3:
• Annual serum prolactin and insulin-like growth factor 1 starting at the age of 5 years
• Annual fasting total serum calcium and/or ionized calcium and PTH starting at the age of 8 years
• Annual fasting serum gastrin starting at the age of 20 years; consider chromogranin A, glucagon, and proinsulin for other enteropancreatic tumors
• Annual fasting glucose starting at the age of 5 years
• Brain magnetic resonance imaging (MRI) at the age of 5 years, repeat every 3 to 5 years based on biochemical test results
• Abdominal computed tomography or MRI starting at the age of 20 years, repeat every 3 to 5 years based on biochemical test results
Surgical management of MEN1 is complex and controversial given the multifocal and multiglandular nature of the disease and the high risk of tumor recurrence even after surgery. A full review of surgical options is outside the scope of this review, but this topic has been reviewed elsewhere.32,33 Establishing the diagnosis of MEN1 before making surgical decisions and referring affected individuals to a surgeon with experience in treating MEN1 can be critical in preventing unnecessary surgeries or inappropriate surgical approaches.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2
Syndrome Description
MEN2, caused by germline mutations in the RET proto-oncogene, is an autosomal dominant syndrome characterized by medullary thyroid cancer (MTC), PC, and/or HPT. Historically, families were classified into one of the three clinical subtypes, MEN2A, MEN2B, and familial medullary thyroid carcinoma (FMTC) based on the presence or absence of certain endocrine tumors and other phenotypic features. However, there is debate about whether FMTC represents a separate entity or is a variation of MEN2A in which there is a lower lifetime risk and delay in the onset of the extrathyroidal manifestations.34 Incorrect classification of families with MEN2A as having FMTC may result in delayed diagnosis of PC, a disease with significant morbidity and mortality. For this reason, current management recommendations include screening for all three tumors in individuals carrying a germline RET mutation, with the exception of parathyroid screening in MEN2B cases35 (see section on management).
The endocrine tumors in MEN2 are often multifocal and bilateral/multiglandular and present at an early age. MTC is present in up to 95% of mutation carriers, and the age at presentation varies, somewhat depending on the specific mutation. Early diagnosis of MTC is critical, given the poor overall survival for individuals diagnosed with distant metastases.36–38 PCs are present in up to 50% of carriers, and the lifetime risk is also dependent on genotype. Although the PCs in MEN2 rarely metastasize, they can be clinically significant because of intractable hypertension or anesthesia-induced hypertensive crises. Parathyroid abnormalities are the least common finding, occurring in up to 30% of patients. The parathyroid disease in MEN2 can include benign parathyroid adenomas or multiglandular hyperplasia, but is typically asymptomatic or associated with only mild elevations in calcium.39,40
MEN2A is diagnosed clinically by the occurrence of two or more of the specific endocrine tumors in a single individual or in close relatives. MEN2A may also be suspected when MTC occurs at an early age (<50 years) or is bilateral or multifocal even in the absence of family history. Several large series indicate a mutation frequency of 1% to 7% in isolated cases of MTC.40,41 Based on these data, it is widely recommended that RET gene mutation testing be performed for all cases of MTC, regardless of age at diagnosis and family history.3,35,42
MEN2B, which makes up 5% of MEN2 cases, is characterized by the early development of an aggressive form of MTC in all patients.43 Patients with MEN2B who do not undergo thyroidectomy at an early age (~1 year) are likely to develop metastatic MTC at an early age. PCs occur in about 50% of MEN2B cases, and clinically significant parathyroid disease is very uncommon.44 Individuals with MEN2B can also have distinctive facies with enlarged lips, mucosal neuromas of the lips and tongue, medullated corneal nerve fibers, and an asthenic marfanoid body habitus.44 About 40% of patients have diffuse ganglioneuromatosis of the gastrointestinal tract.45
Genetic Testing
MEN2 is the result of germline mutations in the RET gene, located on chromosome region 10q11.2.46,47 The RET gene is a proto-oncogene encoding a receptor tyrosine kinase with extracellular, transmembrane, and intracellular domains. RET mutations causing MEN2 are activating mutations, resulting in a constitutively activated tyrosine kinase receptor.48
Genetic testing in MEN2 is considered an important part of the management of at-risk family members. As MTC and other tumors can develop in childhood, testing of children who have no symptoms is considered beneficial.35 Timing of RET testing depends largely on the mutation present in the family. Several groups have developed mutation stratification systems to guide clinicians with regard to the appropriate timing of RET testing, prophylactic thyroidectomy, and biochemical screening.3,35 These are based mainly on age at onset, aggressiveness of thyroid disease, and clinical phenotype, but have not been validated as clinical decision-making tools. The original stratification system was developed by the International RET Consortium.3 A newer classification system by the American Thyroid Association35 was published in 2009 (Table 36.2).
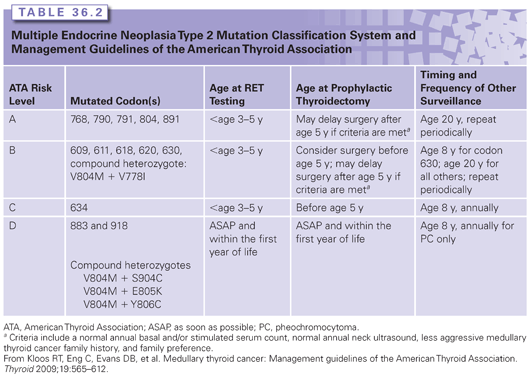
Approximately 95% of patients with MEN2A or MEN2B will have an identifiable germline RET mutation.43 As mentioned previously, 1% to 7% of apparently sporadic cases of MTC will carry a germline RET mutation, underscoring the importance of testing all cases of MTC.3,35,42 A targeted exon approach is most commonly used in families with MEN2A. If the clinical suspicion is MEN2B, a targeted mutation analysis can be used. If targeted testing in a family with a high clinical suspicion for MEN2 is normal, sequencing of the remaining exons can then be performed. For families that do not have a detectable mutation, management recommendations can be based on the clinical features in the affected individual and in the family.
Management
Management of RET mutation carriers includes prophylactic thyroidectomy, as well as biochemical screening for PC and HPT.3,35 The timing of these interventions has been largely based on genotype, but this remains controversial. Prophylactic thyroidectomy and parathyroidectomy with reimplantation of one or more parathyroid glands into the neck or forearm are a preventive option for all subtypes of MEN2. For those with mutations associated with early-onset aggressive MTC, genetic testing alone is used to determine timing of surgery.3,35 For individuals carrying lower- or intermediate-risk mutations (see Table 36.2), some centers allow surgery to be delayed until biochemical screening becomes abnormal and/or the individual reaches a particular age.35 This is still somewhat controversial, however, given the great intrafamilial variability and the fact that MTC can be present even in the absence of an elevated basal or stimulated calcitonin.
Biochemical screening for parathyroid disease and PC is recommended, and the timing and frequency depend on the genotype and, in some cases, presence/absence of these tumors in the family (see Table 36.2). Annual screening for HPT should include albumin-corrected calcium or ionized serum calcium with or without intact PTH measurement (American Thyroid Association, 2009). Screening for PC with plasma-free metanephrines and/or urinary fractionated metanephrines is recommended, given that these provide a higher diagnostic sensitivity than urinary catecholamines.49,50 When biochemical screening suggests PC, MRI or computed tomography can be performed.51,52 Confirmation of the diagnosis can be made using various anatomic and functional modalities.52–54 Several reviews provide a succinct summary of the biochemical diagnosis, localization, and management of PC.52,55 If surgical removal is required, laparoscopic adrenalectomy is the recommended approach for the treatment of unilateral PC.56 For individuals with bilateral PC, cortical-sparing adrenalectomy is an option to minimize the risk of adrenal insufficiency.56,57
PHOSPHATASE AND TENSIN HOMOLOG ON CHROMOSOME 10
Clinical Features
Germline mutations in the PTEN (phosphatase and tensin homolog on chromosome 10) gene have been associated with a number of related clinical disorders, including Cowden syndrome (CS), Bannayan–Riley–Ruvalcaba syndrome, a Proteus-like syndrome, adult Lhermitte–Duclos disease, and autism-like disorders with macrocephaly. Although CS is the only one with a documented risk for endocrine (thyroid) cancer, it is possible that any person with a PTEN mutation is at increased risk.58
The prevalence of CS, an autosomal dominant disorder, has been estimated to be between 1/200,000 and 1/250,000.59 Diagnostic criteria for CS were initially developed in 1996,60 and subsequent modifications have been proposed.61–63 A clinical diagnosis requires a requisite number of clinical features, which are divided into groups of “pathognomonic,” “major,” and “minor” criteria (Table 36.3).

Cancer rates in CS have historically been reported as 25% to 50% for breast cancer, 3% to 10% for thyroid cancer, and 5% to 10% for endometrial cancer, based on compilations of cases published in the early literature.64–66 Thyroid cancer in CS is exclusively of follicular or papillary histology. More recently, an increased risk for colon cancer has been reported.67 Although recent reports from two large cohorts found cancer rates similar to these,67,68
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


