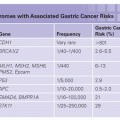
SELECTED GENES THAT MAY CAUSE PANCREATIC CANCER
BRCA2
The BRCA2 gene is associated with hereditary breast and ovarian cancer syndrome, and often presents as premenopausal breast cancer, ovarian cancer, and/or male breast cancer. The Breast Cancer Linkage Consortium5 reported a 3.5-fold (95% confidence interval [CI], 1.9 to 6.6) increased risk of pancreatic cancer in BRCA2 gene mutation carriers. Subsequent studies in the United Kingdom and the Netherlands showed a relative risk of 4.1 and 5.9, respectively.6,7 In a United States–based study, 10.9% (17 out of 156) of families with a BRCA2 mutation reported a family history of pancreatic cancer. The median ages at diagnosis for males and females were 67 and 59 years, respectively, which differed statistically from the Surveillance, Epidemiology and End Results (SEER) database (70 years old for males and 74 years old for females; p = 0.011).8 Although genotype–phenotype data remain sparse, the BRCA2 K3326X variant was found in 5.6% (8 out of 144) of familial pancreatic cancer patients compared with 1.2% (3 out of 250) of those with sporadic pancreatic cancer (odds ratio [OR], 4.84; 95% CI, 1.27 to 18.55; p <0.01).9
Approximately 17% of pancreatic cancer patients who have at least two additional relatives with pancreatic cancer carry deleterious mutations in the BRCA2 gene.10 Estimates for the prevalence of BRCA2 mutations with two first-degree relatives with pancreatic cancer are 6% to 12%,11,12 and BRCA2 mutations also explain a portion of apparently sporadic pancreatic cancers.13 However, prevalence varies between populations. Out of 145 Ashkenazi Jews with pancreatic cancer, 6 (4.1%) were found to have a deleterious BRCA2 mutation when compared with cancer-free controls (OR, 3.85; 95% CI, 2.1 to 10.8; p = 0.007), although no differences were noted in age at diagnosis or clinical pathologic features.14 An earlier, smaller study found a deleterious BRCA2 mutation in 3 (13%) of 23 Ashkenazi Jews with pancreatic cancer, unselected for family history.15 Among Ashkenazi Jewish probands with breast cancer who reported a family history of pancreatic cancer, 7.6% (16 out of 211) had a BRCA2 mutation.16 By comparison, no BRCA2 mutations were found in studies of pancreatic cancer in Korea or Italy.17,18
BRCA1
Similar to BRCA2, mutations in BRCA1 are associated with a markedly increased risk for premenopausal breast cancer and ovarian cancer. The Breast Cancer Linkage Consortium reported a 2.26-fold (95% CI, 1.26 to 4.06) increased risk of pancreatic cancer in families with a BRCA1 mutation,19 and Brose et al.20 estimated a threefold higher lifetime risk. However, more recently, in the United Kingdom, Moran et al.6found no elevation in pancreatic cancer risk in 268 families with a known BRCA1 mutation. A United States–based study reported that 11% (24 out of 219) of their families with a BRCA1 mutation had at least one individual with pancreatic cancer, with median ages at diagnosis of 59 years for males and 68 years for females. Again, this was significantly younger than reported in the SEER database (p = 0.0014).8 Al-Sukhni et al.21 molecularly evaluated pancreatic tumors from seven known BRCA1 mutation carriers and found a loss of heterozygosity of BRCA1 in five (71%), with confirmed loss of the wild-type allele in three of the five compared with only one (11%) of nine sporadic controls. This suggests that BRCA1 germ-line mutations do, in fact, predispose some individuals to pancreatic cancers.
Familial breast cancer registries in the United States and Israel have evaluated the mutation status of families that reported pancreatic cancer in addition to breast cancer and ovarian cancer. In the US study of 19 families with breast, ovarian, and pancreatic cancer, 15 carried a deleterious mutation in BRCA1 and 4 in BRCA2,22 whereas the Israeli study reported an equal number of BRCA1 and BRCA2 families.23
Another study, specifically of Ashkenazi Jewish families, reported a BRCA1 mutation in 7% of probands with breast cancer who also had a family history of pancreatic cancer,16 which was, again, equal to the prevalence of BRCA2 mutations. Thus, within the Ashkenazi Jewish population, BRCA1 and BRCA2 mutations may contribute more equally to risk in families with both breast and pancreatic cancer. However, these studies all examined cohorts of families selected because of clustering of breast and/or ovarian cancer with pancreatic cancer. When families were selected on the basis of familial pancreatic cancer alone, BRCA1 mutations were less prevalent. None of the sixty-six families with three or more cases of pancreatic cancer had a deleterious BRCA1 mutation, including those who also reported a family history of breast and/or ovarian cancer.24 An evaluation of Ashkenazi Jewish patients ascertained on the basis of pancreatic cancer alone showed a 1.3% (2 out of 145) prevalence of BRCA1 mutations.14 Therefore, BRCA1 may explain a small subset of families showing a clustering of pancreatic cancer with breast and/or ovarian cancer, but is unlikely to explain most families with site-specific pancreatic cancer.
PALB2
PALB2 (partner and localizer of BRCA2) was recognized as the FANCN gene in 2007, and biallelic mutation carriers develop Fanconi anemia.25,26 Monoallelic mutation carriers were shown to be at an increased risk for breast cancer (relative risk [RR], 2.3; 95% CI, 1.4 to 3.9).27 The prevalence of PALB2 mutations among familial breast cancer cases is low across ethnicities; PALB2 mutations are relatively nonexistent in breast cancers in the Irish and Icelandic populations and are found in approximately 1% of Italians, African Americans, Chinese, and Spanish breast cancer families, and in 2% of young South African breast cancer patients.28–35 An analysis of 1,144 US familial breast cancer cases found a PALB2 mutation in 3.4% (33 out of 972) of non-Ashkenazi Jews and none (0 out of 172) of Ashkenazi Jews. The estimated risk for breast cancer was 2.3-fold by the age of 55 years (95% CI, 1.5 to 4.2) and 3.4-fold by the age of 85 years (95% CI, 2.4 to 5.9). There was also a fourfold risk for male breast cancer (p = 0.0003) and a sixfold risk for pancreatic cancer (p = 0.002).36 Among French Canadian women with bilateral breast cancer, a PALB2 mutation was found in 0.9% (5 out of 559) compared with none of the 565 women with unilateral breast cancer (p = 0.04), and first-degree relatives of PALB2 mutation carriers had a 5.3-fold risk for breast cancer (95% CI, 1.8 to 13.2).37
PALB2 founder mutations have been identified in several populations, including the c.2323C>T (Q775X) mutation in French Canadians.38 Another example is the Finnish founder mutation c.1592delT. This mutation was found in 2.7% (3 out of 113) of familial breast and/or breast/ovarian cancer families compared with 0.2% (6 out of 2,501) of controls (OR, 11.3; 95% CI, 1.8 to 57.8; p = 0.005).39 One percent (18 out of 1,918) of breast cancer cases unselected for family history also had this founder mutation. The hazard ratio for breast cancer was estimated at 6.1 (95% CI, 2.2 to 17.2; p = 0.01), with a penetrance of 40% by the age of 70 years.40
PALB2 has not been shown to be a significant contributor to familial clustering of other cancers, including melanoma, ovarian cancer, and prostate cancer,41–43 but has been identified in familial pancreatic cancer kindreds. Specifically, Jones et al.44 identified a PALB2 mutation in a familial pancreatic cancer proband, and subsequently found PALB2 mutations in 3 out of 96 additional families, suggesting that 3% to 4% of familial pancreatic cancer may be attributed to this gene. Other populations have found lower mutation frequencies, ranging from absent in the Dutch (0 out of 31) to 3.7% (3 out of 81) in Germans.45,46 When ascertained on the basis of co-occurrence of breast and pancreatic cancer in the same individual or family, prevalence varied, again, from absent in the Dutch (0 out of 45) and United States–based studies (0 out of 77) to 4.8% (3 out of 62) in Italians.42,47,48
CDKN2A
The p16 transcript of the CDKN2A gene is an important cell cycle regulator. Germ-line mutations in the CDKN2A gene predispose individuals to multiple early-onset melanomas. Somatic CDKN2A mutations are also frequently identified in pancreatic adenocarcinomas and precursor lesions, indicating a role for this gene in pancreatic cancer development and progression.49–51
The risk of pancreatic cancer with CDKN2A mutations varies based on genotype. In a study of 22 families with the Dutch founder mutation, p16-Leiden, which is a 19–base-pair deletion in exon 2, the relative risk of pancreatic cancer was 47.8 (95% CI, 28.4 to 74.7).52 The age-related risks have been shown to be less than 1%, 4%, 5%, 12%, and 17% by ages 40, 50, 60, 70, and 75 years, respectively.53 Regarding other mutations, the Genes, Environment and Melanoma Study assessed relative risks for nonmelanoma cancers in 429 first-degree relatives of 65 melanoma patients with a CDKN2A mutation. Five pancreatic cancers were reported compared with 41 pancreatic cancers among 23,452 first-degree relatives of 3,537 noncarriers, for a relative risk of 7.4 (95% CI, 2.3 to 18.7; p = 0.002).54 A United States–based study estimated penetrance to be 58% by the age of 80 years (95% CI, 8% to 86%) and noted a hazard ratio of 25.8 (p = 2.1 × 10213) in those who ever smoked cigarettes.55
Mutation prevalence in pancreatic cancer families varies by population. In an Italian study, 5.7% of 225 consecutive patients with pancreatic cancer had an identified CDKN2A mutation.56 The predominant mutations were the E27X and G101W founder mutations, although others were also represented. Of 16 patients classified as having familial pancreatic cancer, 5 (31%) carried CDKN2A
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


