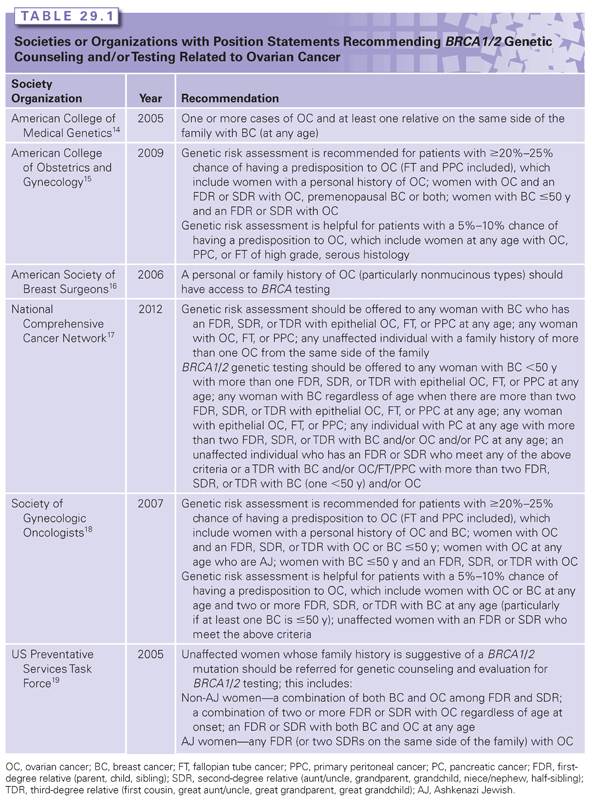
HEREDITARY BREAST AND OVARIAN CANCER SYNDROME (THE BRCA1 AND BRCA2 GENES)
Hereditary breast and ovarian cancer syndrome due to mutations in the BRCA1 and BRCA2 (BRCA1/2) genes is the most common cause of hereditary ovarian cancer, including fallopian tube and primary peritoneal cancer. Anywhere from 0.125% to 0.20% of the general population carry mutations in BRCA1/2 compared with 15% of women with a diagnosis of invasive ovarian cancer.6,8–10 The contribution of the BRCA1/2 genes to ovarian cancer is even greater in certain ethnicities that have higher BRCA1/2 mutation prevalence rates (e.g., Polish, Ashkenazi Jewish [AJ], French Canadian). For example, the prevalence of BRCA1/2 mutations in individuals of AJ ancestry and women of AJ ancestry with ovarian cancer is ~2.3% and ~30% to 40%, respectively.11–13 Because of the strong connection between ovarian cancer and BRCA1/2, multiple professional societies and organizations recommend genetic counseling and testing for any woman with ovarian cancer regardless of age at onset or family history (Table 29.1). The current sensitivity of BRCA1/2 genetic testing at the commercial laboratory is ~90% for identifying mutations in either gene.
The lifetime risk of developing ovarian cancer differs between the two genes. A number of studies over the years have quantified the lifetime risk20–26; however, two large meta-analyses suggest an ~40% and 20% lifetime risk for BRCA1 and BRCA2 mutation carriers, respectively.27,28 In 1997, Gayther et al.,29 in an effort to determine whether there were any genotype–phenotype correlations associated with BRCA2 mutations, identified a region in exon 11 of BRCA2 between nucleotides 3,035 and 6,629 that appeared to confer a further increased risk of ovarian cancer; they coined this region the “ovarian cancer cluster region” (OCCR). A follow-up study of the OCCR in families from the Breast Cancer Linkage Consortium (which included the families from the study by Gayther et al.29) further refined the OCCR to nucleotides 3,059 to 4,075 and 6,503 to 6,629, but found that the phenotype of increased ovarian cancer may actually be due to a reduced breast cancer risk.30 Regardless, the potential difference in ovarian cancer risk was not significant enough to affect management recommendations. In addition to genotype–phenotype associations, researchers have studied other genetic modifiers (e.g., single-nucleotide polymorphisms) that can influence ovarian cancer risk in BRCA1/2 mutation carriers, but these data need maturation before testing for genetic modifiers can be used clinically.31–34
The average age at onset of ovarian cancer is between 49 and 53 years for BRCA1 and 55 and 58 years for BRCA2 compared with 63 years in the general population.6,8,35–37 Unlike breast cancer, women with a diagnosis of very early-onset ovarian cancer (>40 years) are significantly less likely to harbor BRCA1/2 mutations.8,26,35,38,39 This is in part due to the fact that early-onset ovarian cancers are more likely to be associated with borderline tumors, earlier stages, and more favorable histologic characteristics, none of which are typical of BRCA1/2-related ovarian cancer.40
BRCA1/2-associated ovarian cancers are almost uniformly epithelial in origin and, for the most part, are invasive and nonmucinous; there are case reports of germ cell and stromal tumors in BRCA1/2 mutation carriers. Mucinous and borderline tumors individually account for ~2% of ovarian tumors identified in BRCA1/2 mutation carriers; this percentage is the same for both prospective and retrospective analyses, which are nicely summarized by Evans et al.41 Compared with sporadic ovarian cancers, BRCA1/2 ovarian cancers are more often of serous histology, higher grade, and solid type, and have intact p53 staining on immunohistochemistry (IHC).39,41–44 It is important to note that other histologic findings are seen in mutation carriers (e.g., endometrioid, clear cell, papillary), and one study found more giant cell–type cancers in BRCA1 mutation carriers compared with controls.42 Several smaller studies have found that BRCA1/2-related ovarian cancers have a better prognosis compared with ovarian cancers in nonmutation carriers.39,45–48 This finding seems to have been confirmed as a recent large pooled analysis of 26 observational studies comparing 3,879 BRCA1/2 ovarian cancers and 2,666 noncarriers found the 5-year survival for BRCA2 carriers was 52%, 44% for BRCA1 carriers, and 36% for noncarriers.44 The survival difference remained after adjusting for age at diagnosis, stage, histology, and grade.
One potential reason for the differences in survival may be that BRCA1/2-related ovarian cancers respond better to platinum-based agents.45,48 BRCA1/2 repair DNA damage through homologous recombination and platinum agents are particularly active in cells deficient in homologous recombination.48,49 It is through this pathway that another class of drugs called poly(ADP-ribose) polymerase (PARP) inhibitors has been developed to help treat BRCA1/2-related cancers. Unlike platinum agents targeting homologous recombination, PARP inhibitors block repair of single-strand DNA breaks through base excision repair, which in turn can lead to double-strand breaks that cannot be repaired by BRCA1/2-deficient tumor cells at the same time sparing normal cells.50–52 A number of phase 1 and phase 2 trials have been reported, and clinical trials continue to study PARP inhibitors in both ovarian and breast cancers.53–56
Identifying women who have a BRCA1/2 mutation would ideally lead to women either being diagnosed with ovarian cancer at earlier stages or preventing ovarian cancer altogether. When counseling women who have tested positive for a BRCA1/2 mutation, it is these central tenets, ovarian cancer screening versus prevention, that guide discussions. Current National Comprehensive Cancer Network® Guidelines for managing ovarian cancer risk include the recommendation for risk-reducing bilateral salpingo-oophorectomy (RRSO) between the ages of 35 and 40 years when childbearing is complete; for women not choosing RRSO, transvaginal ultrasound and CA-125 are recommended every 6 months starting at age 30 years or 5 to 10 years before the earliest age at onset of ovarian cancer in the family.17 Surgical prevention is recommended over screening for two main reasons. First, many women can receive a dual risk reduction with one surgery. RRSO has been shown to reduce ovarian cancer risk by 80% to 95%,57–60 and breast cancer risk by 50% (for premenopausal women) in BRCA1/2 mutation carriers.59–62 Anywhere from 2.5% to 17%63–65 of women who undergo RRSO are found to have an occult ovarian, fallopian tube, or primary peritoneal cancer upon pathology review, emphasizing the need for a “high-risk” pathologic examination of the tissues.66 However, after surgery, women may still face an <4% risk of developing primary peritoneal cancer over the course of 20 years.57 The second major reason surgery is recommended over screening is that ovarian cancer screening is ineffective at detecting ovarian cancer at early stages67; the pros and cons of ovarian cancer screening with transvaginal ultrasound and CA-125 have been reviewed elsewhere.4 A second option for reducing ovarian cancer risk is through oral contraceptive (OCP) use. OCPs use can reduce ovarian cancer risk by <50% with 5 years of use; the benefit has been shown for both BRCA1 and BRCA2 carriers.68,69 However, it is worth noting that data are conflicting about whether OCP use can increase breast cancer risk, so it is important to take into account a woman’s age, family history, and genetic test results before making recommendations to use OCPs.70–73 Recently, researchers at Stanford created an online decision tool to aid female BRCA1/2 mutation carriers in conjunction with a health-care provider making decisions with respect to cancer screening and surgical prevention74; the tool is available at http://brcatool.stanford.edu.
LYNCH SYNDROME
Approximately 2% to 4% of ovarian cancer is believed to be associated with Lynch syndrome, also referred to as hereditary nonpolyposis colorectal cancer syndrome.7,75 Lynch syndrome is an autosomal dominant cancer predisposition syndrome characterized by a significantly increased lifetime risk of colorectal cancer (~30% to 70%) and extracolonic malignancies of the endometrium (28% to 60%), stomach (6% to 9%), small bowel (3% to 4%), urinary tract (3% to 8%), central nervous system (4%), hepatobiliary tract (1%), and sebaceous skin lesions (1% to 9%).76
Lynch syndrome is caused by germline mismatch repair (MMR) gene mutations in MLH1, MSH2, MSH6, and PMS2. Alterations in these four genes account for approximately 36%, 38%, 14%, and 15% of Lynch syndrome, respectively.77 In addition, germline deletions in EPCAM inactivate MSH2 through epigenetic silencing in a small proportion of individuals with Lynch syndrome.78 Variations in cancer risk have been noted among the four MMR genes. An increased risk for endometrial cancer and slightly decreased risk for colorectal cancer are observed in families with germline MSH6 mutations compared with families with MLH1 and MSH2 mutations.79 Germline mutations in PMS2 are associated with the lowest overall risk for Lynch syndrome–associated malignancies.80
Lynch syndrome is diagnosed clinically within families meeting either the Amsterdam I or Amsterdam II criteria81,82 (Table 29.2). Although fulfillment of the Amsterdam criteria is a significant predictor of identifying a germline MMR gene mutation in a family, it is well known that at least 25% of families affected with Lynch syndrome do not meet the Amsterdam criteria.83

The lifetime risk of ovarian cancer in Lynch syndrome is estimated to be anywhere between 4% and 11%,78,84,85 with a mean age of diagnosis at 42.7 years.75 In this series, approximately one-third of the individuals were younger than 40 years at the time of diagnosis (n = 80). Approximately 94% of ovarian tumors in Lynch syndrome are invasive epithelial in origin, with borderline and granulosa cell tumors representing ~4% of cases. Lynch-related ovarian tumors are most frequently moderately or well differentiated. Of note, synchronous endometrial cancer was identified in 21.5% of cases. In a meta-analysis of 159 cases of MMR-related ovarian cancers, histologic subtypes included serous (32%), endometrioid (29%), mixed (24%), mucinous (19%), and clear cell (18%).87 In a study of individuals with Lynch-related ovarian cancer compared with individuals with sporadic ovarian cancer, no significant difference was observed in survival rate, although the total number of Lynch-related ovarian cancers was small (n = 277).87
Lynch syndrome is one of a few hereditary cancer syndromes that have more than one method available to make a genetic diagnosis by identifying MMR deficiency including (1) tumor studies, specifically microsatellite instability (MSI) and IHC, and (2) germline genetic testing (typically includes DNA sequencing and a technology to look for large structural rearrangements). In initiating a genetic evaluation for Lynch syndrome, tumor studies are generally the recommended first-line tests.78 However, because the majority of available data on performing MSI and IHC pertain to colorectal and endometrial tumors,76–78 it is not certain whether tumor studies on ovarian cancers as a first-line approach are valid. Approximately 12% of unselected ovarian cancers will have an MSI-high phenotype.75 In an analysis of 52 ovarian carcinomas in a population who received a diagnosis at younger than 50 years, defects in MMR expression were identified in 10% of cases using MSI and IHC.88 Domanska et al.89 evaluated ovarian carcinomas in a population who received a diagnosis at younger than 40 years and found MMR deficiency with IHC in ~6% of cases. If tumor studies are indicative of Lynch syndrome (results show the tumor to be MSI-high and/or loss of ≥1 MMR proteins), germline genetic testing should be offered and will identify a deleterious mutation anywhere from 20% to 70% of the time.78 Molecular analysis is widely available for the four MMR genes and EPCAM. However, if tumor studies are performed on an ovarian cancer, and the results are not indicative of Lynch syndrome, given the paucity of data, Lynch syndrome cannot conclusively be ruled out, and germline genetic testing may still be warranted, depending on the patient’s age at onset and family history.
If a tumor specimen is not available, germline genetic testing may be initiated at the outset. A recent abstract showed that up to 4% of unselected ovarian cancers may be found to have a germline mutation in MLH1, MSH2, or MSH6.7 If a patient with ovarian cancer needs a genetic evaluation for Lynch syndrome, the ideal approach would be for the health-care provider to have a detailed discussion about the pros and cons of each testing methodology with the patient, so that the patient and provider can jointly come to a decision about the best approach given the rest of the family history of cancer.
Current ovarian cancer management guidelines for women with Lynch syndrome include prophylactic hysterectomy and bilateral salpingo-oophorectomy after completion of childbearing; there is not enough data to support routine ovarian cancer screening with transvaginal ultrasound and CA-125 blood tests for women, although it may be considered at the clinician’s discretion.90
PEUTZ–JEGHERS SYNDROME
Peutz–Jeghers syndrome (PJS) is a rare autosomal dominant inherited disorder characterized by gastrointestinal hamartomatous polyposis, mucocutaneous melanin pigmentation, and benign and malignant tumors of the gastrointestinal tract, breast, ovary, cervix, and testis. The incidence of PJS is unknown but has been estimated between 1:8,300 and 1:200,000 births.91,92
In contrast to the other hamartomatous syndromes in which polyps occur most commonly in the colon, PJS-related polyps occur most commonly in the small intestine (90%), although they can also occur elsewhere in the gastrointestinal tract including the stomach (25%) and large bowel (33%), and may also develop outside the digestive tract in the uterus, bladder, lungs, and nasal passages.93
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


