Management
The current recommendations for the screening of women at risk for HBOC is based on the best available evidence and is expected to change as more specific features of BRCA1– and BRCA2-related disease become available. The current screening recommendations for women are listed in Table 28.2.

Risk-reduction mastectomies are an appropriate consideration for women at the highest hereditary risk for breast cancer. Studies have shown a 90% to 95% reduction in breast cancer risk following prophylactic mastectomy.44–47 The evidence for the use of tamoxifen or raloxifene as a chemopreventive agent in BRCA carriers is limited; however, tamoxifen has been shown to reduce the risk of contralateral breast cancers in BRCA carriers.48,49 Two recent studies support the role of risk-reducing salpingo-oophorectomy: The hazard ratio for ovarian cancer for women who underwent prophylactic surgery and that for those who chose close surveillance were 0.15 and 0.04, respectively.50,51 Women should be informed about the potential for the subsequent development of peritoneal carcinomatosis, which has been reported up to 15 years after risk-reducing bilateral salpingo-oophorectomy.25,52 Combination oral contraceptives containing estrogen and progestin result in a protective effect against ovarian cancer in some studies, but not in others.53–55
Male BRCA mutation carriers are advised to undergo training in breast self-examination with regular monthly practice and semiannual clinical breast examinations, and workup of any suspected breast lesions is recommended. The NCCN Clinical Practice Guidelines In Oncology (NCCN Guidelines®) also recommend that a baseline mammogram be considered, with an annual mammogram if gynecomastia or parenchymal/glandular breast density is identified on baseline study.56 The NCCN Guidelines® recommend that male BRCA mutation carriers should adhere to the current prostate cancer screening guidelines.56,57
Psychosocial Considerations
The psychosocial needs of BRCA-positive women have been studied fairly widely. Studies have shown that although there is slight worsening of distress symptoms following cancer genetic counseling in BRCA1/BRCA2 mutation carriers, these symptoms were minimal, did not affect everyday life activities, and had almost disappeared at 1-year follow-up.58–62 Approximately 20% of BRCA1/2 mutation carrier women experience high distress after learning their test result.63,64 Factors that are related to high posttest distress include a high level of pretest anxiety, higher pretest perceived risk, and whether they are opting for prophylactic surgery to reduce their risk.5 It is important to note, however, that even in women who experienced distress after receipt of genetic test information, women do not “regret” their decision to be tested.66 It has been suggested that health-care providers consider including a brief pretest psychological assessment before initiating genetic testing for BRCA1 and BRCA267 so that these women can be targeted for more comprehensive support once test results are available.68
The anxiety-associated symptoms reported by BRCA1/2 carriers include sleeplessness and “bad mood.”60,69,70 One other psychosocial issue reported by single women with BRCA1/2 mutations is that they experience increased urgency at finding a life partner capable of handling the emotional strain of the cancer world and open to pursuing multiple paths toward parenthood.71
Various studies have suggested that existing social support networks are inadequate for BRCA1/2 mutation carriers and that formal services are unavailable or underutilized.66,70,72 To address this lack of formal support services, a retreat for BRCA1/2 carriers that includes educational updates about medical management, genetic privacy, and discrimination and addresses psychological and family issues may provide a valuable opportunity for BRCA carriers and their families to receive updated medical information, share personal experiences, provide and receive support, and change health behaviors.73
Distress in male BRCA carriers has not be studied quite as widely, but one study noted that high distress after disclosure of the result was reported by one of four male mutation carriers.74
TP53
Description
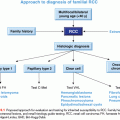
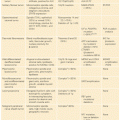
Germline mutations in the TP53 gene give rise to a disease called Li-Fraumeni syndrome (LFS), which is a rare cancer predisposition syndrome thought to be responsible for ~1% of breast cancers.75 LFS is often thought of as a hereditary predisposition to cancer in general, involving many tumor types and occurring at any point in an individual’s lifetime, including childhood. The majority of cases of LFS are due to mutations in the p53 gene.76–79 The component tumors of LFS include bone sarcomas (primarily osteosarcomas and chondrosarcomas), soft tissue sarcomas, breast cancer, brain tumors, leukemia, and adrenocortical carcinomas.80 The classic component tumors are thought to account for 63% to 77% of cancer diagnoses in individuals with LFS.80–83 Breast cancer is the most common tumor in p53 mutation carriers (24% to 31.2%), followed by soft tissue sarcomas (11.6% to 17.8%), brain tumors (3.5% to 14%), osteosarcomas (12.6% to 13.4%), and adrenocortical tumors (6.5% to 9.9%).84,85 Other tumors that have been argued to be component tumors of LFS are listed in Table 28.3.
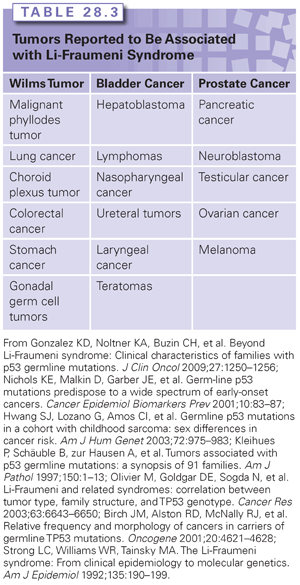
There are some data regarding common histology of LFS component tumors. Breast cancers are most commonly invasive ductal carcinomas.80 Rhabdomyosarcomas account for 55% of soft tissue sarcomas, followed by fibrosarcomas (13%) and then malignant fibrous histiocytomas.84 For LFS-associated brain tumors, 69% are astrocytic (astrocytoma or glioblastoma), followed by medulloblastoma/primitive neuroectodermal tumors (17%).84
Identifying Li-Fraumeni Syndrome
Li et al.80 first defined LFS in 1988 at which point clinical criteria were established, now known as classic LFS criteria (Table 28.4). In 1994, Birch et al.77 went on to define less stringent criteria (see Table 28.4) in an attempt to capture families with p53 mutations who did not necessarily conform to the classic criteria. Families who met the broader criteria of Birch et al.77 were referred to as “LFS-like (LFL)” families. Both classic and LFL criteria are based on family history and fail to recognize potential p53 mutation carriers who have de novo germline p53 mutations. Although the de novo rate is not well defined for p53, one study showed as high as a 24% rate.88

More recently in 2001, Chompret et al.89 developed criteria for identifying patients likely to carry p53 mutations (see Table 28.4) and included criteria that address families who display a collection of component tumors but also address individuals whose personal histories are suggestive of p53 mutation even in the absence of a suggestive family history. The Chompret criteria were designed to include individuals who may potentially carry de novo p53 mutations.
Fifty to seventy percent of individuals who meet the classic definition of LFS will have a mutation in p53.77,89–92 Individuals who meet the LFL criteria are less likely to be p53 mutation carriers, estimated at 21% to 40%.79,90 Twenty percent of individuals meeting the Chompret criteria will be identified as p53 mutation carriers.89
Cancer Risks
Typically, LFS-associated tumors occur at significantly younger ages than when they occur sporadically. However, depending on tumor type, the mean age at diagnosis varies from childhood well into adulthood.84 Understanding cancer risk for LFS is somewhat complicated as the ranges of risk vary greatly between studies and depend largely on study population. When pooling studies that examine overall cancer risk in p53 mutation carriers (both female and male), the risk of developing cancer by ages 15 to 20 years is 12% to 42%, by ages 40 to 45 years is 52% to 66%, by age 50 years is 80%, and by age 85 years is 85%.82,83,88,93 When separating out the sexes, it is apparent that female p53 mutation carriers have generally a higher lifetime cancer risk in comparison to males.83,88,94
Individuals with a diagnosis of LFS are also at markedly increased risk of developing multiple primary tumors. Hisada et al.95 found that, following a first cancer diagnosis, there is a 57% risk for a second primary tumor within 30 years of the first diagnosis, followed by a 38% risk for a third primary tumor within 10 years of the second cancer diagnosis. In addition, it has been widely observed that second, third, and so on primary cancers commonly occur in the radiation field of previously treated cancers.76,80,88,95
Psychosocial Issues
The psychosocial effects of being a member of an LFS family and/or being affected with LFS have not been widely studied.96,97 The nature of the disease itself leads to unique psychosocial implications with individual members of LFS families often experiencing many cancer diagnoses (and deaths) in their immediate and extended family. These cancer diagnoses will be throughout the life span, with many parents having to deal with a child’s diagnosis and many children needing to deal with a parent’s diagnosis. It is likely that these repeated experiences of grief and stress pose a significant psychological burden for the members of LFS families.98 Although no data exist, this psychosocial burden may also impact individuals’ relationships with their family members including, but not limited to, children and spouses.
Because of the rarity of the syndrome, many individuals with LFS may feel isolated. Other inherited syndromes, in general, and inherited cancer syndromes, in particular, have “support groups” that can help with the coping process when an individual is diagnosed with the disease. Unfortunately, no such group exists in the United States today. An online discussion group/support group for individuals with LFS is available (http://listserv.acor.org/SCRIPTS/WA-ACOR.EXE?OK=53111E8B&L=LI-FRAUMENI). Members of the listserv include patients with LFS, health-care providers, and spouses and friends of individuals with LFS. The listserv serves as a place not only to share information about the disease but also to discuss fears, anxiety, grief, and other psychological manifestations of the disease.
COWDEN SYNDROME (PHOSPHATE AND TENSIN HOMOLOG)
Description
CS is a rare hereditary cancer syndrome that is characterized by overgrowth in different organ systems. The incidence of CS is thought to be about 1 in 200,000, but it may be underdiagnosed.99 CS belongs to the set of syndromes known as the phosphatase and tensin homolog (PTEN) hamartoma tumor syndromes.100 PTEN mutations are found in the vast majority of patients with CS, although mutations in other genes such as BMPR1A and the succinate dehydrogenase genes have been reported in a small number of patients who have features of CS but do not meet diagnostic criteria (CS-like).101,102
Diagnostic Criteria Testing Criteria
Traditionally, one of the hallmark features of CS is the development of multiple hamartomas of the skin and mucosa. A thorough physical examination, including head circumference measurement and examination for skin manifestations, is an important component of assessing for CS. However, a lack of hamartomas does not exclude CS; diagnostic criteria are complicated.103 The NCCN®’s most recent guidelines (v.2.2014) for testing for CS are in Table 28.5.

Identifying Cowden Syndrome
In 2011, the Cleveland Clinic made available an online calculator for risk of a PTEN mutation in adults, as well as a set of pediatric criteria (http://www.lerner.ccf.org/gmi/ccscore/). Risk estimates were based on data from the largest prospective cohort of patients collected with a potential diagnosis of CS. Information on physical findings, specific cancer diagnoses, intestinal polyps, and other benign conditions is collected. If a patient has a risk of mutation greater than 3%, testing for PTEN is recommended.104
Cancer Risks
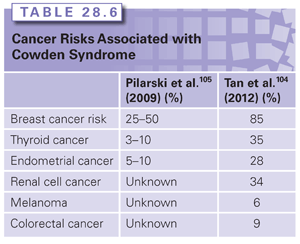
The highest risk of cancer associated with CS is for female breast cancer. Other cancers that are thought to be a part of the spectrum of cancers seen in CS include thyroid cancer and uterine cancer; more recently, renal cell cancer, melanoma, and colorectal cancer have also been reported. The magnitude of risk for the cancers associated with CS varies widely.104,105 A recent article from Cleveland Clinic estimated the lifetime risks of cancer to be much higher than previously reported; however, it is likely that there is significant ascertainment bias present in this cohort.104 A comparison of two publications reviewing the cancer risks associated with CS is presented in Table 28.6.
Management
CS is a complex diagnosis to make and to receive. Because of the degree of variability in CS, it is difficult for clinicians to make a firm diagnosis except in the most obvious of cases. In situations where there is a high suspicion, a negative genetic test result may be uninformative for the patient and her family. Conversely, a positive result or variant of uncertain significance in an individual without classic features of Cowden can lead to uncertainty regarding how aggressive to be about screening and prevention measures. The NCCN Guidelines for management are in Table 28.7.

Psychosocial Issues
There is a dearth of literature addressing the psychological issues for individuals and families with a clinical and/or genetic diagnosis of CS, possibly due to its rarity. However, there are several factors associated with CS that could add to the psychological burden of having a hereditary syndrome. These include variability in clinical presentation, difficulty screening (especially for breast cancer), disfigurement due to mucocutaneous lesions and surgical procedures, the possibility of intellectual disabilities and/or autism in children, lack of knowledge about how often PTEN mutations are found de novo versus inherited in a family, a large number of uncertain variants found through genetic testing, and overall lack of knowledge about the syndrome.
Because of the association of CS with autism and macrocephaly, many children are now undergoing genetic testing for alterations in the PTEN gene; a small number of them will be found to have CS or a related disorder.106 When the child is the index case in the family, testing him/her may provide information for adult family members about cancer risks. In addition, parents may find value in knowing that there is an underlying genetic cause to their child’s issues and in finding a community with a shared diagnosis. There is also the hope that the development of targeted therapies may help ameliorate the disease in children and, going forward, in adults.
The benefit of testing an asymptomatic child whose parent has a known mutation in PTEN is still unknown. Although childhood cancers have been reported in CS, these cancers appear to be rare. Some experts argue that thyroid and other screening is warranted in children for the early detection and prevention of related cancers107
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


