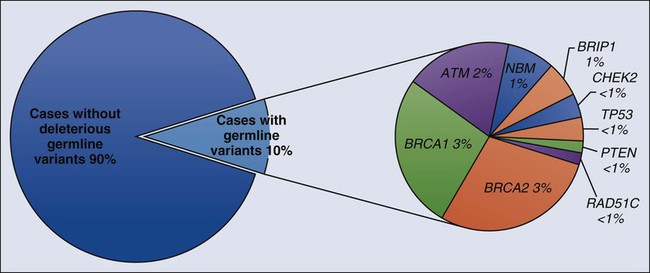
12 Kasmintan A. Schrader, Ravi Sharaf, Shaheen Alanee and Kenneth Offit • The discovery of inherited mutations of genes associated with increased risk for cancer provides important clinical opportunities for early detection and prevention of common and rare forms of human malignancies. • Syndromes of cancer predisposition often involve multiple organ systems, affect paired organs with bilateral or multifocal tumors, and have onset at an earlier age compared with nonfamilial tumors. The diagnosis of particular cancer predisposition syndromes can usually be confirmed with molecular genetic testing of patients who have hereditary malignancies. Genetic testing can then be extended to relatives as a predictive test to guide their preventive management. • Medical, surgical, and radiation oncologists, genetic counselors, and allied professionals are playing a leading role in the integration of genetic testing into the practice of preventive oncology. Recently, genomic analysis has been applied to tumors to discover targets for therapy. Because tumor genomic analysis will also include a comparison with the germline, the need for genetic counseling for both cancer and noncancer disease risks will be increased. This chapter reviews both common and more recently described familial cancer syndromes, with an emphasis on the clinical application of cancer genetic and genomic analysis in the management of patients who have or are at risk for cancer. During the past decade, the availability of clinical testing for inherited mutations of cancer predisposition genes has had a major impact on the practice of clinical oncology.1 As these genes were identified and characterized, guidelines for the responsible clinical translation of this information were developed by medical and surgical subspecialty societies, for example, the statements of the American Society of Clinical Oncology in 19962 and 20033 and related educational materials.6–6 These guidelines emphasized that in the process of offering a predictive genetic test to a patient or family that is affected by cancer, the provider and the individual who is being tested must be prepared to deal with all the medical, psychological, and social consequences of a positive, negative, or ambiguous result. These guidelines define the form and content of genetic counseling as a component of cancer risk assessment and management. A selected set of syndromes of cancer predisposition, listed in Table 12-1, is reviewed in this chapter. More detailed discussions of breast, colon, gastric, pancreatic, and renal cancer susceptibility are found in the chapters that discuss these tumors. A more comprehensive list of syndromes is provided in Table 12-2. Whether offered by a physician, genetic counselor, or other health care professional, genetic testing for inherited cancer risk requires careful informed consent. The elements of informed consent for genetic testing are summarized in Box 12-1. With the recent advent of genomic screening of tumors, as well as gene panel testing and genomic screening in the germline, these elements of informed consent must also include the eventuality of “incidental” findings associated with risk of cancer or noncancer diseases.7,8 Table 12-1 Selected Cancer Predisposition Syndromes Table 12-2 Syndromes of Inherited Cancer Predisposition in Clinical Oncology *OMIM, On-Line Mendelian Inheritance in Man, <http://www.ncbi.nlm.nih.gov/Omim/>; 2013 [accessed 12.02.13]. Mutations (or pathogenic changes) in genes whose alterations result in susceptibility to cancer may be inherited, whereby a family history usually includes multiple individuals diagnosed with cancer, or they can occur seemingly sporadically (or de novo) in families in which the history of cancer may be unremarkable. In many instances, the same genes found to be recurrently mutated in specific sporadic cancers, if mutated in the germline, are also susceptibility genes for the same types of tumors. This phenomenon reflects the genotype-phenotype relationships that arise as a result of aberration of particular biological pathways and thus define the patterns that characterize hereditary cancer syndromes (see Table 12-2). It is now widely accepted that cancer is a disease of the genome, whereby particular genetic changes (“drivers”) set the stage for abnormal cellular function, resulting in uncontrolled cell division and generally loss of the normal fidelity of DNA repair and replication. This process results in accumulation of further mutations, which either contribute to tumor survival or are merely “passengers” (or collateral damage) in the neoplastic process. With the advent of massively parallel sequencing, a rapid gain in the understanding of tumor genetics has occurred, whereby these “driver” and “passenger” mutations are now being identified and cataloged. The importance of identifying the “driver” mutations, their genes, and respective biological pathways not only relate to the development of therapies targeted toward relevant pathways that likely are integral for that tumor’s survival, but also with regard to prognostication and the potential relevance to cancer susceptibility. Inherited cancer predisposition should be considered as a spectrum, arising from single or combined low-, moderate-, and high-risk genetic variants for which the timing of disease onset is likely modified by the type of genetic variant and its effect on normal cellular function and the responses to environmental factors. Highly penetrant hereditary cancer syndromes generally account for about 5% to 10% of most types of cancer and are caused by rare genetic variants. Genes associated with hereditary cancer syndromes have traditionally been found by studying the DNA of large families with multiple affected individuals and linking the disease phenotype to regions of the genome to determine candidate genes from within the linkage region. These genes are then sequenced to look for a causative mutation. Although linkage studies were successful in identifying most of the known hereditary cancer syndromes, a large proportion of recognized familiality within certain cancer types remained unaccounted for and was thought to be due to more common inherited factors. This situation led to large-scale association studies of common genetic factors (polymorphisms) across the entire genomes of thousands of unrelated persons with more common types of cancer, resulting in lists of single nucleotide polymorphisms that marginally increase or decrease risk for these cancers,9 although unexplained excess familiality still remained. In the wake of new and more efficient sequencing technologies, the focus has shifted back toward studying families segregating multiple cases of cancer. Sequencing of all of the protein coding regions of the genome or the entire genome itself has led to the discovery of novel cancer susceptibility genes (Box 12-2). Together the knowledge of rare and common variation has the potential to inform the clinician about the combined effect of genetic factors that lead to cancer, extending beyond risks conferred by single genes. Ultimately, assessing a person’s risk of cancer will relate to understanding his or her inherent combination of genetic factors within the context of environmental exposures, which also may include exposures to cancer therapies. The major syndromes of cancer predisposition that affect adults include common syndromes associated with breast, ovarian, colon, gastric, and pancreatic cancer, as well as a number of other less common but equally important cancer predispositions. Some of these syndromes, including multiple endocrine neoplasia type I, retinoblastoma, melanoma, and von Hippel–Lindau (VHL) disease, are described elsewhere in this text. Comprehensive reviews of these cancer predispositions are offered elsewhere, including multiple endocrine neoplasia type I,10,11 retinoblastoma,12 melanoma,13,14 and VHL disease.15,16 Some syndromes such as neurofibromatosis also have cancer predispositions.19–19 A detailed outline of the elements of setting up a comprehensive cancer genetic and genomics counseling service has also been published and provides the sources of Table 12-2.1 Although only about 18,000 cases of breast cancer each year are associated with an obvious hereditary predisposition, primary cancers developed in more than 200,000 breast cancer survivors in the United States as a result of a hereditary predisposition, and these survivors remain at risk for secondary cancers.20, 21 Genetic testing has emerged as one of the most important indicators of risk factors pointing to a need for intensified screening for breast cancer.22 When detected at an early stage, more than 90% of breast cancers are curable. These statistics underscore the rationale for the use of genetics in clinical oncology. We have previously reviewed in detail the management of women and men who are at hereditary risk for breast cancer,23 and this review will be summarized and updated here. From 1 in 150 to 1 in 800 individuals in the population carry a genetic susceptibility to breast cancer,26–26 and the prevalence is much higher in certain ethnic groups. Syndromes of breast cancer susceptibility are linked to mutations of BRCA1 and BRCA2, as well as a smaller number of cases with germline mutations of RAD51C, PTEN, p53, CHEK2, STK11, CDH1, and rarer syndromes (Table 12-3). Table 12-3 Known Genes Associated with Hereditary Breast Cancer Predisposition BC, Breast cancer; HBOC, hereditary breast and ovarian cancer syndrome. aAntoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003;72:1117–30. bFord D, Easton DF, Bishop DT, et al. Risks of cancer in BRCA1-mutation carriers: Breast Cancer Linkage Consortium. Lancet 1994;343:692–5. cOffit K. BRCA mutation frequency and penetrance: new data, old debate. J Natl Cancer Inst 2006;98:1675–7. dSatagopan JM, Offit K, Foulkes W, et al. The lifetime risks of breast cancer in Ashkenazi Jewish carriers of BRCA1 and BRCA2 mutations. Cancer Epidemiol Biomarkers Prev 2001;10:467–73. eLalloo F, Varley J, Ellis D, et al. Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet 2003;361:1101–2. fChen S, Iversen ES, Friebel T, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 2006;24:863–71. gBirch JM, Alston RD, McNally RJ, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 2001;20:4621–8. hChompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000;82:1932–7. iEvans DG, Birch JM, Thorneycroft M, et al. Low rate of TP53 germline mutations in breast cancer/sarcoma families not fulfilling classical criteria for Li-Fraumeni syndrome. J Med Genet 2002;39:941–4. jBrownstein MH, Wolf M, Bikowski JB. Cowden’s disease: a cutaneous marker of breast cancer. Cancer 1978;41:2393–8. kStarink TM, van der Veen JP, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet 1986;29:222–33. lZbuk KM, Stein JL, Eng C: PTEN hamartoma tumor syndrome (PHTS), <http://www.genetests.org/servlet/access?db=geneclinics&site=gt&id=8888891&key=cVUld9gO6ESTy&gry=&fcn=y&fw=XU2v&filename=/profiles/phts/index.html>; 2013 [accessed 12.02.13]. mGiardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53. nHearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006;12:3209–15. oYoon KA, Ku JL, Yang HK, et al. Germline mutations of E-cadherin gene in Korean familial gastric cancer patients. J Hum Genet 1999;44:177–80. pBrooks-Wilson AR, Kaurah P, Suriano G, et al. Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J Med Genet 2004;41:508–17. qPharoah PD, Guilford P, Caldas C, et al. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001;121:1348–53. rAthma P, Rappaport R, Swift M. Molecular genotyping shows that ataxia-telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet Cytogenet 1996;92:130–4. sBerstein JL, Concannon P, Langholz B, et al. Multi-center screening of mutations in the ATM gene among women with breast cancer: the WECARE Study. Radiat Res 2005;163:698–9. tBretsky P, Haiman CA, Gilad S, et al. The relationship between twenty missense ATM variants and breast cancer risk: the Multiethnic Cohort. Cancer Epidemiol Biomarkers Prev 2003;12:733–8. uCHEK2 Breast Cancer Case-Control Consortium. CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet 2004;74:1175–82. vMeijers-Heijboer H, van den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002;31:55–9. wThompson D, Seal S, Schutte M, et al. A multicenter study of cancer incidence in CHEK2 1100delC mutation carriers. Cancer Epidemiol Biomarkers Prev 2006;15:2542–5. xShaag A, Walsh T, Renbaum P, et al. Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Hum Mol Genet 2005;14:555–63. ySeal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 2006;38:1239–41. zRahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 2007;39:165–7. aaPark DJ, Lesueur F, Nguyen-DumontT, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet 2012;90:734–9. Recently, germline mutations in RAD51C have been identified in families with breast and ovarian cancer.27,28 Follow-up studies indicate that RAD51C is a low-frequency, moderate- to high-risk ovarian cancer susceptibility gene with the relative risk for ovarian cancer in RAD51C-mutation carriers as high as fivefold.31–31 Cowden syndrome (CS) was initially described as a dominant inheritance of multiple hamartomatous lesions, including papillomas of the lips and mucous membranes and acral keratoses of the skin.32 This disease was ultimately linked to germline mutations of PTEN. In persons with Li-Fraumeni syndrome, early-onset breast cancer occurs with soft-tissue sarcomas, osteosarcoma, leukemia, brain tumors, adrenal cortical tumors, and other cancers. Rarely, a typical breast-ovarian kindred may be found to have a germline p53 mutation.33 In Northern European families, specific mutations of CHEK2 are associated with familial breast cancer.34 Although the common European CHEK2 mutation is rare in persons in North America,35 analysis of women for CHEK2 founder mutations stratified for family history of breast cancer demonstrated that carriers with a positive family history had a greater than 25% lifetime risk for breast cancer.36 Women with Peutz-Jeghers syndrome carry germline mutations in the STK11 gene and are at increased risk for breast cancer.37,38 Both benign and malignant breast tumors occur in Muir-Torre syndrome, a variant of hereditary nonpolyposis colon cancer that is associated with germline mutations of MSH2 and MLH1. Carriers of ATM mutations have ~twofold elevated breast cancer risk.41–41 Early linkage studies suggested that in about 50% of breast cancer kindreds, the cancer was linked to BRCA1; in 30%, it was linked to BRCA2; and in the remainder, it was linked to BRCA3 and other as yet unidentified genes.42 In up to two thirds of families with both male and female breast cancer, the cancers were due to BRCA2, whereas more than 80% of families with both breast and ovarian cancer harbored BRCA1 mutations.43 Multiple, common, lower penetrance genes are likely to account for a significant component of currently unexplained familial breast cancer risk.44 Two such low-penetrance breast cancer alleles that were revealed in large population studies are BRIP1 and PALB2.47–47 A host of putative lower penetrant gene mutations have been identified through whole-genome association studies,48,49 although the clinical relevance of these associations remains unclear. Recently, two rare variants in XRCC2 have been identified as causing breast cancer susceptibility.50 An analysis of the tumor exomes of 507 unselected breast tumors revealed ~10% to have underlying germline mutations of genes, including ATM, BRCA1, BRCA2, BRIP1, CHEK2, NBN, PTEN, RAD51, and p53 (Fig. 12-1).51 Compared with the 10% breast cancer risk for women in the general population, estimates of the breast cancer risk that is conferred by a common susceptibility gene ranged from 67% to 69% by age 70 years based on epidemiological analyses.24,25 Genetic linkage studies of families that were selected because of early-onset breast or ovarian cancer gave risk estimates as high as 70% to 90% in families in which mutations of either of these two genes were segregated.54–54 Ovarian cancer risks in these families varied from 10% to 80%, and risk for a second breast cancer was as high as 64% by age 70 years. More recent estimates based on population studies led to slightly lower risk estimates, with a lifetime breast cancer risk of 56% by age 70 years (confidence interval: 40% to 73%), and ovarian cancer risk estimated at 16%.55 Penetrance estimates that were determined through clinic-based ascertainment of families revealed a 64% risk for breast or ovarian cancer by age 70 years.56 The role of ascertainment and other possible biases in deriving these estimates, as well as risks for cancer of the prostate, colon, pancreas, and other sites in BRCA mutation carriers has been reviewed.57 In addition to Fanconi anemia, persons with compound BRCA2 mutations may experience childhood medulloblastomas.58 BRCA1 and BRCA2 are both important for DNA repair, in particular homologous recombination, which enables repair of double-stranded DNA breaks. BRCA1 is a large gene, spanning more than 100,000 bases of genomic DNA with 22 coding and two noncoding exons. BRCA2 is also large, consisting of 27 exons across 70 kb of genomic DNA. Both genes, by coincidence, have a large exon 11. An update of reported mutations is accessible through the Internet at http://research.nhgri.nih.gov/bic/. Most BRCA1 mutations cause premature truncation of the peptide by frameshift or nonsense sequence changes. Large germline rearrangements also occur in BRCA1 and BRCA2.59, 60 Five percent to 10% of BRCA mutations that are missense are problematic because they are of unknown clinical significance. The proportion of these variants that are of unknown significance was as high as 10% to 23% in some series, posing counseling challenges.61,62 The role of BRCA1 and BRCA2 in DNA damage response and homologous recombination is reviewed elsewhere.63 Founder BRCA mutations have been documented in genetically isolated populations. In North American families, the most common founder mutations occur in persons of Ashkenazi Jewish origin. These mutations include a two-base-pair deletion in codon 23 of BRCA1, termed 185delAG (c.68_69delAG); another BRCA1 mutation, 5382insC (c.5266dupC); and the 6174delT (c.5946delT) mutation in BRCA2.63–66 About 1 in 40 Ashkenazi Jews harbor one of the common BRCA1 or BRCA2 mutations,55,56,65 a relatively high carrier frequency for an inherited cancer predisposition syndrome. Other mutations in BRCA1 and BRCA2 occur in the Ashkenazim; 16 of 737 Ashkenazi Jews who were tested in a clinic-based ascertainment had a nonfounder mutation (2%).43 In another study of Ashkenazi individuals with a personal history of breast or ovarian cancer who had previously been shown not to have a founder mutation, 3 of 70 (4.3%) had a deleterious nonfounder mutation.67 Founder mutations in populations other than the Ashkenazim have also been observed.70–70 BRCA1-linked tumors are associated with a basal-like subtype and higher mitotic indices.71 They were of higher grade, were more frequently estrogen and progesterone receptor negative,72,73 and demonstrated a basal-like phenotype.74 They displayed a more “aggressive” phenotype, including a higher proportion of cells in S phase, and other indices.73,75–78 The prognostic significance of BRCA mutations is still being defined. Epithelial ovarian carcinoma can be delineated into five distinct histologic subtypes: high-grade serous, clear cell, endometrioid, mucinous, and low-grade serous with differing manifestations, prognoses, molecular profiles, and immunohistochemistry profiles.81–81 The high-grade serous subtype comprises the majority of cases of ovarian cancer.80 Mutations in BRCA1 and BRCA2 show a strong association with the high-grade serous carcinoma of ovary/fallopian tube or peritoneal origin, such that germline testing for BRCA1 and BRCA2 together can have up to a 22.6% to 25% percent detection rate in this histologic subtype.82–85 Furthermore, massively parallel sequencing of 360 subjects with ovarian, fallopian tube, or peritoneal cancer for 21 tumor suppressor genes revealed germline loss-of-function mutations in 24%: 18% in BRCA1 or BRCA2 and, additionally, 6% in BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C, or TP53.28 Three elements of breast surveillance that are recommended to BRCA heterozygotes are self-examination, clinician examination, and imaging. The evidence base underlying these recommendations has been reviewed,23,86 and updated guidelines are available on the Web.87 Increasingly, breast cancer screening including mammography and magnetic resonance imaging (MRI) together have been shown to have greatest sensitivity.88 For women who are at the highest hereditary risk for breast cancer, whose breasts are difficult to examine or who have had biopsies showing atypia, it might be appropriate to discuss the option of removing the healthy breasts as a preventive measure (i.e., prophylactic mastectomy). Retrospective studies and reviews document the well-established risk-reducing role of surgery in high-risk patients.91–91 Prospective and combined consortium studies have also shown the efficacy of this approach.34,92, 93 Because of their antiestrogen properties, tamoxifen, raloxifene, and a newer class of synthetic estrogen receptor modulators emerged as hormonal chemopreventive agents that have been shown to decrease breast cancer rates in persons who are at increased risk.94 The safety of these drugs in premenopausal women remains to be established. Two studies on the impact of tamoxifen on subsequent breast cancer risk in BRCA1 and BRCA2 mutation carriers have shown conflicting conclusions,95,96 although only one of these studies was sufficiently powered to reach significant results. Tamoxifen was confirmed to decrease contralateral breast cancer risk in a follow-up of one of these studies of BRCA mutation carriers.97 Small trials have demonstrated the ability of ultrasound with Doppler and CA125 to find early-stage ovarian cancers in BRCA mutation carriers.98,99 However, the efficacy of this approach has not been proven in large, prospective studies. Therefore prophylactic removal of the ovaries and fallopian tubes is recommended to women with strong family histories of ovarian cancer, in families linked to BRCA1 or BRCA2, or to women who are considering hysterectomy in the setting of a germline mutation associated with Lynch syndrome. Studies examining specimens from prophylactic salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers implicate the fimbriated part of the fallopian tube as harboring precursor lesions to high-grade serous epithelial ovarian cancer.100–103 Thus prophylactic salpingo-oophorectomy is recommended because such surgeries may not only decrease the incidence of subsequent breast and ovarian cancer but also find occult early-stage pelvic neoplasms,102,104,105 decreasing mortality.106 These studies have also confirmed that serous surface carcinoma, also called papillary serous carcinoma, can still occur after prophylactic oophorectomy,107,108 leading most authors to use the term risk-reducing salpingo-oophorectomy. Combination oral contraceptives that contain estrogen and high-dose progestin result in a time-dependent, protective effect against ovarian cancer in some but not all studies of BRCA mutation carriers.111–111 There remains the concern of a small increased risk of breast cancer due to oral contraceptives in this group, particularly in persons with BRCA1 mutations.112 Germline BRCA1 and BRCA2 mutation-associated tumors have defective DNA repair mechanisms because of the loss of function of the remaining normal copy of the gene. This inability of the tumor to repair its DNA effectively can be exploited by therapies that disrupt alternative DNA repair pathways, such that the tumor cell accumulates so much DNA damage that it can no longer survive.113,114 Inhibitors of the base excision repair enzyme Poly(adenosine diphosphate-ribose) polymerase utilize this principle of synthetic lethality and are currently in clinical trials.115 These drugs may also have a role in cancers associated with germline mutations in BRCA2, including cancer of the pancreas (relative risk [RR] 4.1, 95% confidence interval [CI] 1.9-7.8), prostate (RR 6.3, 95% CI 4.3-9.0), and uveal melanoma (RR 99.4, 95% CI 11.1-359.8).116 CS is an autosomal-dominant disorder characterized by multiple hamartomas with a high risk of benign and malignant tumors of the thyroid, breast, and endometrium. Consensus criteria for CS establish three diagnostic categories.117 Pathognomonic criteria include adult Lhermitte-Duclos disease (LDD) (defined as presence of a cerebellar dysplastic gangliocytoma)118 and mucocutaneous lesions, facial trichilemmomas, acral keratoses, papillomatous lesions, and mucosal lesions. Major criteria include breast cancer, thyroid cancer (especially follicular histology), macrocephaly,119 and endometrial carcinoma. Minor criteria include other thyroid lesions (e.g., goiter), mental retardation, hamartomatous intestinal polyps, fibrocystic breast disease, lipomas, fibromas, and genitourinary tumors (e.g., uterine fibroids and renal cell carcinoma) or genitourinary malformation. The operational diagnosis of CS is made if an individual meets any one of the following criteria: pathognomonic mucocutaneous lesions alone if there are six or more facial papules, of which three or more must be trichilemmoma, or cutaneous facial papules and oral mucosal papillomatosis; or oral mucosal papillomatosis and acral keratoses; or six or more palmoplantar keratoses. Alternatively, the individual may fulfill two major criteria, but one must have either macrocephaly or LDD. Alternatively, the individual may have one major and three minor criteria or four minor criteria. The palmar and plantar hyperkeratotic pits usually become evident later in childhood. Subcutaneous lipomas and cutaneous hemangiomas are seen in persons with CS with low frequency.120 An increased risk of early-onset male breast cancer has been noted in mutation carriers.121 Cumulative lifetime risks for female breast cancer are 81% to 85.2%122,123; for LDD, 32% (CI: 19% to 49%)122; for thyroid cancer, 21% to 35.2%,122,123; for endometrial cancer, 19% to 28.2%122,123; and for renal cancer, 15% to 33.6% (CI: 6% to 32%).122, 123 The lifetime risks for colorectal cancer are 9% to 16%,122, 123 and the risk for melanoma is 6%.123 The CS-linked PTEN was mapped to 10q22-23.124 Bannayan-Riley-Ruvalcaba syndrome, characterized by macrocephaly, intestinal polyps, lipomas, and pigmented penile macules, is also caused by germline mutations in PTEN.125 PTEN acts as a tumor suppressor by mediating cell cycle arrest, apoptosis, or both.126 Full sequencing and deletion/duplication analysis are available clinically, and promoter analysis is available on a research basis. Heterozygous germline mutations in PTEN cause most cases of CS. Nonsense, missense, and frameshift mutations that are predicted to disrupt normal PTEN function have been identified in some families,124 including mutations that disrupt the protein tyrosine/dual-specificity phosphatase domain. Initially it was shown that PTEN mutations can be detected in about 80% of patients with CS.125 More recently the mutation detection rate in persons meeting CS criteria was found to be 34%, raising the possibility that current aspects of testing criteria are too broad.127 Persons with known germline PTEN mutations should undergo appropriate cancer screening.87,128 In view of recent work outlining cancer risks in persons with CS, some investigators have suggested that National Comprehensive Cancer Network management guidelines be modified to include renal cancer screening using biannual renal imaging from age 40 years or 5 years earlier than the earliest kidney cancer diagnosis in the family, and endometrial sampling in adulthood or 5 years earlier than the earliest endometrial cancer diagnosis in the family.123 Female patients with CS should have breast self-examination training and education starting at age 18 years. Beginning at age 25 years, clinical breast examinations should be performed every 6 to 12 months, and annual mammography and MRI screening should start at age 30 years or 5 years before the earliest age of breast cancer diagnosis in the family.123 Men should perform monthly breast self-examination. Female patients should receive endometrial cancer screening beginning around age 30 years or 5 years before the earliest age of endometrial cancer diagnosis in the family.123 Persons with CS should undergo biannual colonoscopy from age 40 years.123 Both men and women should have a comprehensive annual physical examination starting at diagnosis with screening for skin and thyroid lesions, including a baseline and annual thyroid ultrasound and dermatologic examination.123
Genetic Factors
Hereditary Cancer Predisposition Syndromes
Syndrome
Gene
Hereditary breast and ovarian cancer
BRCA1, BRCA2
Cowden syndrome
PTEN
Lynch syndrome
MSH2, MLH1, MSH6,PMS2
Familial adenomatous polyposis
APC
MUTYH-associated polyposis
MUTYH
Hereditary diffuse gastric cancer
CDH1
Carney complex
PRKAR1A
Hereditary paraganglioma-pheochromocytoma syndromes
SDHB, SDHC, SDHD, SDHAF2
Multiple endocrine neoplasia (type II)
RET
Gorlin syndrome
PTCH
Hereditary leiomyomatosis and renal cell cancer syndrome
FH
von Hippel–Lindau disease
VHL
Birt-Hogg-Dubé syndrome
FLCN
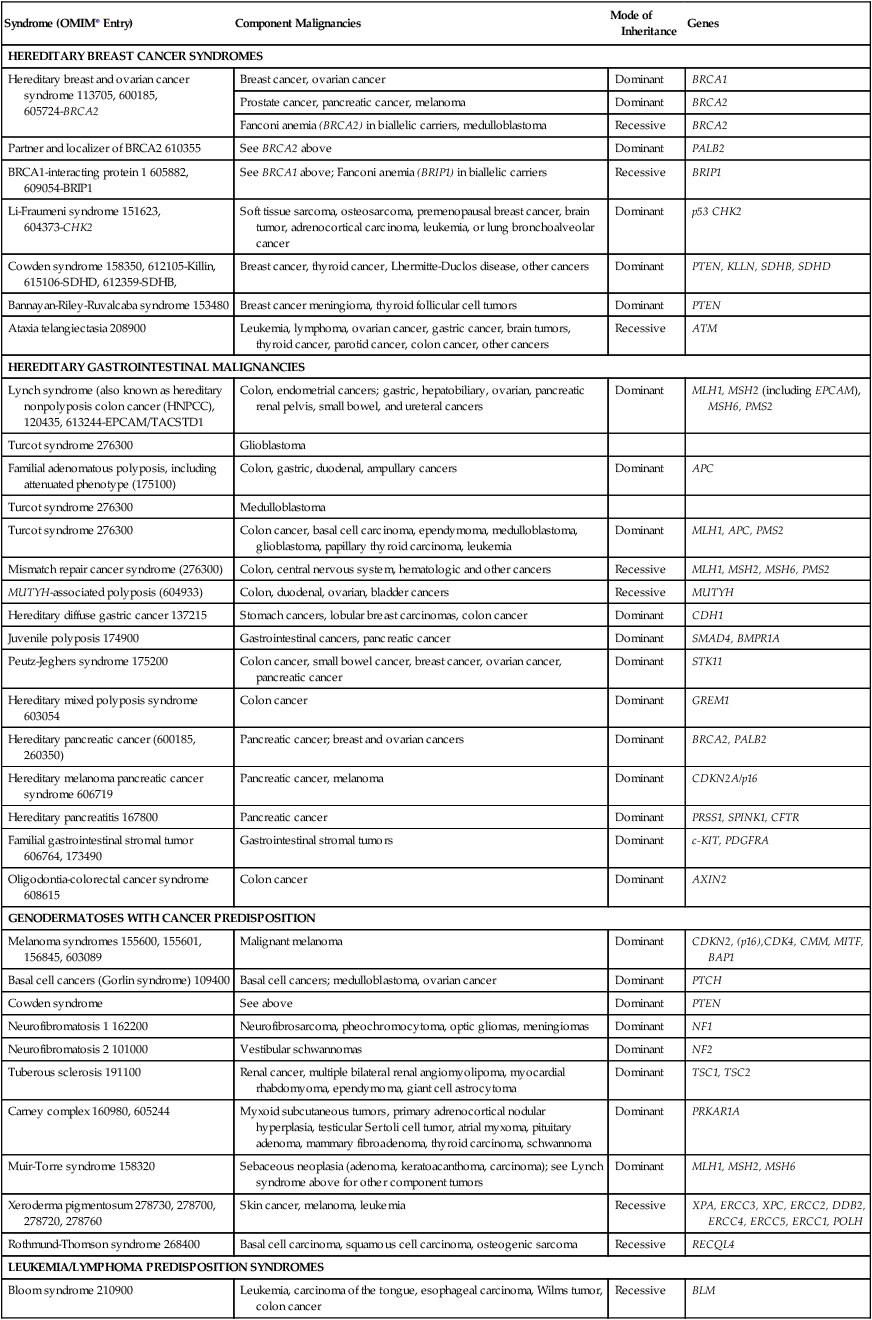
Syndrome (OMIM* Entry)
Component Malignancies
Mode of Inheritance
Genes
HEREDITARY BREAST CANCER SYNDROMES
Hereditary breast and ovarian cancer syndrome 113705, 600185, 605724-BRCA2
Breast cancer, ovarian cancer
Dominant
BRCA1
Prostate cancer, pancreatic cancer, melanoma
Dominant
BRCA2
Fanconi anemia (BRCA2) in biallelic carriers, medulloblastoma
Recessive
BRCA2
Partner and localizer of BRCA2 610355
See BRCA2 above
Dominant
PALB2
BRCA1-interacting protein 1 605882, 609054-BRIP1
See BRCA1 above; Fanconi anemia (BRIP1) in biallelic carriers
Recessive
BRIP1
Li-Fraumeni syndrome 151623, 604373-CHK2
Soft tissue sarcoma, osteosarcoma, premenopausal breast cancer, brain tumor, adrenocortical carcinoma, leukemia, or lung bronchoalveolar cancer
Dominant
p53 CHK2
Cowden syndrome 158350, 612105-Killin, 615106-SDHD, 612359-SDHB,
Breast cancer, thyroid cancer, Lhermitte-Duclos disease, other cancers
Dominant
PTEN, KLLN, SDHB, SDHD
Bannayan-Riley-Ruvalcaba syndrome 153480
Breast cancer meningioma, thyroid follicular cell tumors
Dominant
PTEN
Ataxia telangiectasia 208900
Leukemia, lymphoma, ovarian cancer, gastric cancer, brain tumors, thyroid cancer, parotid cancer, colon cancer, other cancers
Recessive
ATM
HEREDITARY GASTROINTESTINAL MALIGNANCIES
Lynch syndrome (also known as hereditary nonpolyposis colon cancer (HNPCC), 120435, 613244-EPCAM/TACSTD1
Colon, endometrial cancers; gastric, hepatobiliary, ovarian, pancreatic renal pelvis, small bowel, and ureteral cancers
Dominant
MLH1, MSH2 (including EPCAM), MSH6, PMS2
Turcot syndrome 276300
Glioblastoma
Familial adenomatous polyposis, including attenuated phenotype (175100)
Colon, gastric, duodenal, ampullary cancers
Dominant
APC
Turcot syndrome 276300
Medulloblastoma
Turcot syndrome 276300
Colon cancer, basal cell carcinoma, ependymoma, medulloblastoma, glioblastoma, papillary thyroid carcinoma, leukemia
Dominant
MLH1, APC, PMS2
Mismatch repair cancer syndrome (276300)
Colon, central nervous system, hematologic and other cancers
Recessive
MLH1, MSH2, MSH6, PMS2
MUTYH-associated polyposis (604933)
Colon, duodenal, ovarian, bladder cancers
Recessive
MUTYH
Hereditary diffuse gastric cancer 137215
Stomach cancers, lobular breast carcinomas, colon cancer
Dominant
CDH1
Juvenile polyposis 174900
Gastrointestinal cancers, pancreatic cancer
Dominant
SMAD4, BMPR1A
Peutz-Jeghers syndrome 175200
Colon cancer, small bowel cancer, breast cancer, ovarian cancer, pancreatic cancer
Dominant
STK11
Hereditary mixed polyposis syndrome 603054
Colon cancer
Dominant
GREM1
Hereditary pancreatic cancer (600185, 260350)
Pancreatic cancer; breast and ovarian cancers
Dominant
BRCA2, PALB2
Hereditary melanoma pancreatic cancer syndrome 606719
Pancreatic cancer, melanoma
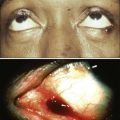
Dominant
CDKN2A/p16
Hereditary pancreatitis 167800
Pancreatic cancer
Dominant
PRSS1, SPINK1, CFTR
Familial gastrointestinal stromal tumor 606764, 173490
Gastrointestinal stromal tumors
Dominant
c-KIT, PDGFRA
Oligodontia-colorectal cancer syndrome 608615
Colon cancer
Dominant
AXIN2
GENODERMATOSES WITH CANCER PREDISPOSITION
Melanoma syndromes 155600, 155601, 156845, 603089
Malignant melanoma
Dominant
CDKN2, (p16),CDK4, CMM, MITF, BAP1
Basal cell cancers (Gorlin syndrome) 109400
Basal cell cancers; medulloblastoma, ovarian cancer
Dominant
PTCH
Cowden syndrome
See above
Dominant
PTEN
Neurofibromatosis 1 162200
Neurofibrosarcoma, pheochromocytoma, optic gliomas, meningiomas
Dominant
NF1
Neurofibromatosis 2 101000
Vestibular schwannomas
Dominant
NF2
Tuberous sclerosis 191100
Renal cancer, multiple bilateral renal angiomyolipoma, myocardial rhabdomyoma, ependymoma, giant cell astrocytoma
Dominant
TSC1, TSC2
Carney complex 160980, 605244
Myxoid subcutaneous tumors, primary adrenocortical nodular hyperplasia, testicular Sertoli cell tumor, atrial myxoma, pituitary adenoma, mammary fibroadenoma, thyroid carcinoma, schwannoma
Dominant
PRKAR1A
Muir-Torre syndrome 158320
Sebaceous neoplasia (adenoma, keratoacanthoma, carcinoma); see Lynch syndrome above for other component tumors
Dominant
MLH1, MSH2, MSH6
Xeroderma pigmentosum 278730, 278700, 278720, 278760
Skin cancer, melanoma, leukemia
Recessive
XPA, ERCC3, XPC, ERCC2, DDB2, ERCC4, ERCC5, ERCC1, POLH
Rothmund-Thomson syndrome 268400
Basal cell carcinoma, squamous cell carcinoma, osteogenic sarcoma
Recessive
RECQL4
LEUKEMIA/LYMPHOMA PREDISPOSITION SYNDROMES
Bloom syndrome 210900
Leukemia, carcinoma of the tongue, esophageal carcinoma, Wilms tumor, colon cancer
Recessive
BLM
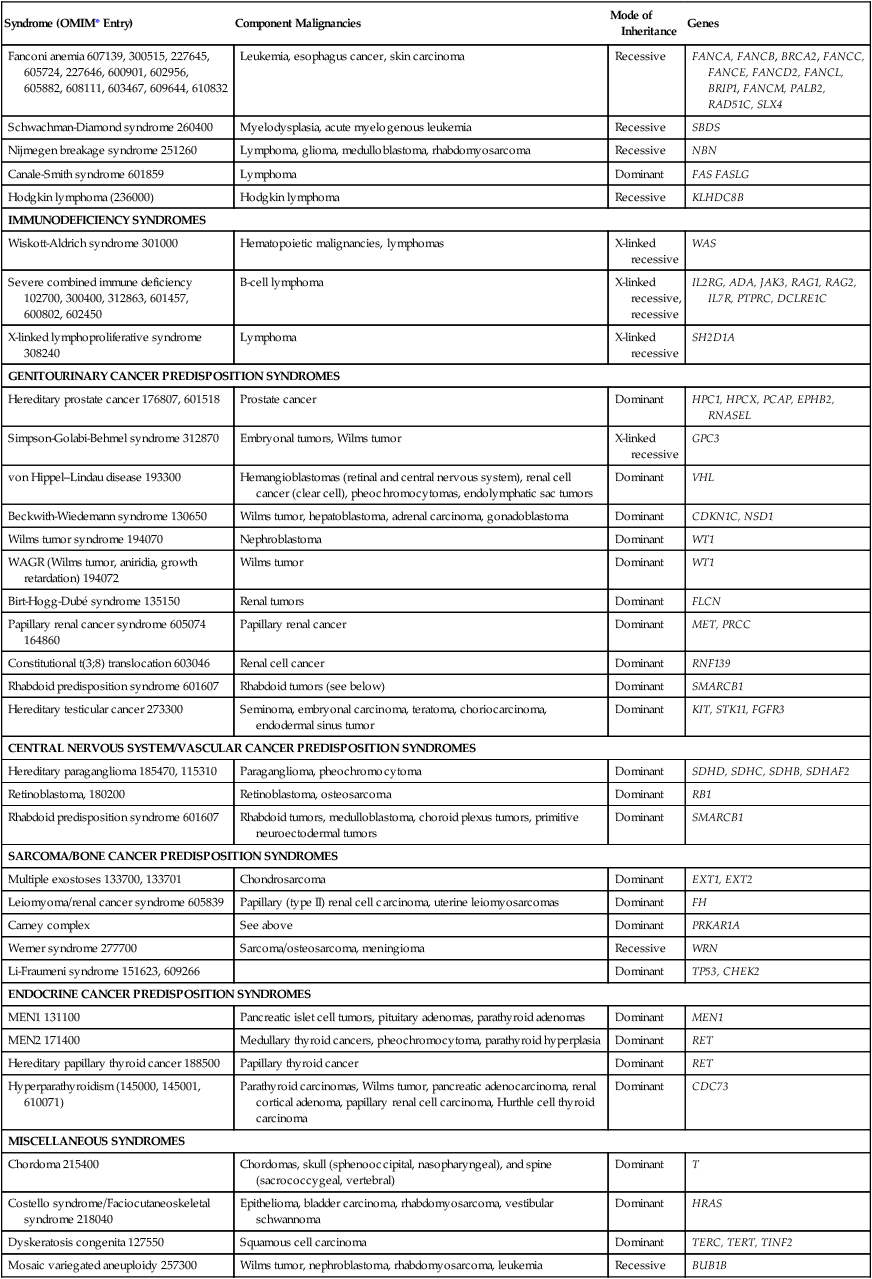
Fanconi anemia 607139, 300515, 227645, 605724, 227646, 600901, 602956, 605882, 608111, 603467, 609644, 610832
Leukemia, esophagus cancer, skin carcinoma
Recessive
FANCA, FANCB, BRCA2, FANCC, FANCE, FANCD2, FANCL, BRIP1, FANCM, PALB2, RAD51C, SLX4
Schwachman-Diamond syndrome 260400
Myelodysplasia, acute myelogenous leukemia
Recessive
SBDS
Nijmegen breakage syndrome 251260
Lymphoma, glioma, medulloblastoma, rhabdomyosarcoma
Recessive
NBN
Canale-Smith syndrome 601859
Lymphoma
Dominant
FAS FASLG
Hodgkin lymphoma (236000)
Hodgkin lymphoma
Recessive
KLHDC8B
IMMUNODEFICIENCY SYNDROMES
Wiskott-Aldrich syndrome 301000
Hematopoietic malignancies, lymphomas
X-linked recessive
WAS
Severe combined immune deficiency 102700, 300400, 312863, 601457, 600802, 602450
B-cell lymphoma
X-linked recessive, recessive
IL2RG, ADA, JAK3, RAG1, RAG2, IL7R, PTPRC, DCLRE1C
X-linked lymphoproliferative syndrome 308240
Lymphoma
X-linked recessive
SH2D1A
GENITOURINARY CANCER PREDISPOSITION SYNDROMES
Hereditary prostate cancer 176807, 601518
Prostate cancer
Dominant
HPC1, HPCX, PCAP, EPHB2, RNASEL
Simpson-Golabi-Behmel syndrome 312870
Embryonal tumors, Wilms tumor
X-linked recessive
GPC3
von Hippel–Lindau disease 193300
Hemangioblastomas (retinal and central nervous system), renal cell cancer (clear cell), pheochromocytomas, endolymphatic sac tumors
Dominant
VHL
Beckwith-Wiedemann syndrome 130650
Wilms tumor, hepatoblastoma, adrenal carcinoma, gonadoblastoma
Dominant
CDKN1C, NSD1
Wilms tumor syndrome 194070
Nephroblastoma
Dominant
WT1
WAGR (Wilms tumor, aniridia, growth retardation) 194072
Wilms tumor
Dominant
WT1
Birt-Hogg-Dubé syndrome 135150
Renal tumors
Dominant
FLCN
Papillary renal cancer syndrome 605074 164860
Papillary renal cancer
Dominant
MET, PRCC
Constitutional t(3;8) translocation 603046
Renal cell cancer
Dominant
RNF139
Rhabdoid predisposition syndrome 601607
Rhabdoid tumors (see below)
Dominant
SMARCB1
Hereditary testicular cancer 273300
Seminoma, embryonal carcinoma, teratoma, choriocarcinoma, endodermal sinus tumor
Dominant
KIT, STK11, FGFR3
CENTRAL NERVOUS SYSTEM/VASCULAR CANCER PREDISPOSITION SYNDROMES
Hereditary paraganglioma 185470, 115310
Paraganglioma, pheochromocytoma
Dominant
SDHD, SDHC, SDHB, SDHAF2
Retinoblastoma, 180200
Retinoblastoma, osteosarcoma
Dominant
RB1
Rhabdoid predisposition syndrome 601607
Rhabdoid tumors, medulloblastoma, choroid plexus tumors, primitive neuroectodermal tumors
Dominant
SMARCB1
SARCOMA/BONE CANCER PREDISPOSITION SYNDROMES
Multiple exostoses 133700, 133701
Chondrosarcoma
Dominant
EXT1, EXT2
Leiomyoma/renal cancer syndrome 605839
Papillary (type II) renal cell carcinoma, uterine leiomyosarcomas
Dominant
FH
Carney complex
See above
Dominant
PRKAR1A
Werner syndrome 277700
Sarcoma/osteosarcoma, meningioma
Recessive
WRN
Li-Fraumeni syndrome 151623, 609266
Dominant
TP53, CHEK2
ENDOCRINE CANCER PREDISPOSITION SYNDROMES
MEN1 131100
Pancreatic islet cell tumors, pituitary adenomas, parathyroid adenomas
Dominant
MEN1
MEN2 171400
Medullary thyroid cancers, pheochromocytoma, parathyroid hyperplasia
Dominant
RET
Hereditary papillary thyroid cancer 188500
Papillary thyroid cancer
Dominant
RET
Hyperparathyroidism (145000, 145001, 610071)
Parathyroid carcinomas, Wilms tumor, pancreatic adenocarcinoma, renal cortical adenoma, papillary renal cell carcinoma, Hurthle cell thyroid carcinoma
Dominant
CDC73
MISCELLANEOUS SYNDROMES
Chordoma 215400
Chordomas, skull (sphenooccipital, nasopharyngeal), and spine (sacrococcygeal, vertebral)
Dominant
T
Costello syndrome/Faciocutaneoskeletal syndrome 218040
Epithelioma, bladder carcinoma, rhabdomyosarcoma, vestibular schwannoma
Dominant
HRAS
Dyskeratosis congenita 127550
Squamous cell carcinoma
Dominant
TERC, TERT, TINF2
Mosaic variegated aneuploidy 257300
Wilms tumor, nephroblastoma, rhabdomyosarcoma, leukemia
Recessive
BUB1B
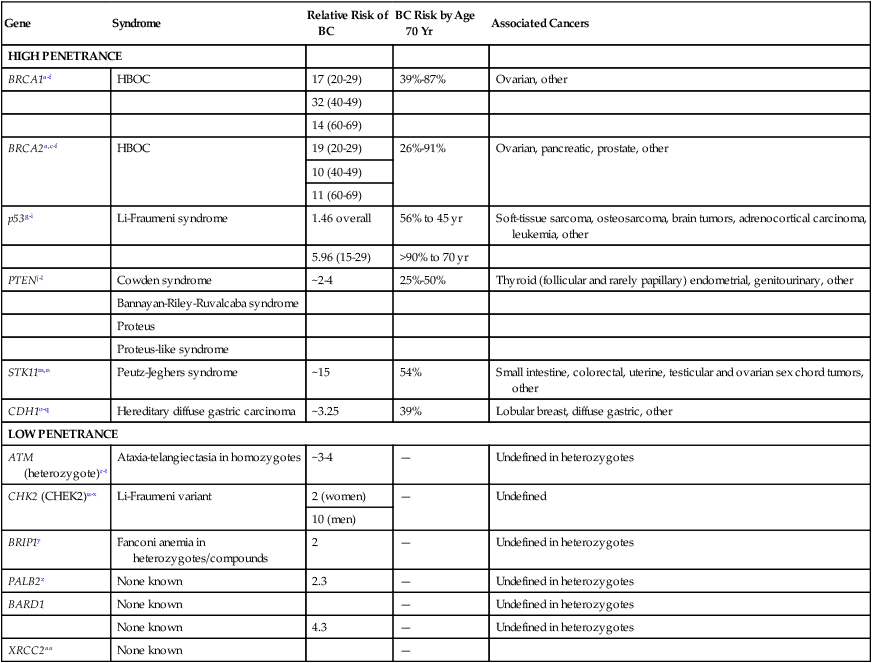
Common Syndromes of Cancer Predisposition
Breast and Ovarian Cancer Syndromes
Clinical Features
Gene
Syndrome
Relative Risk of BC
BC Risk by Age 70 Yr
Associated Cancers
HIGH PENETRANCE
BRCA1a–f
HBOC
17 (20-29)
39%-87%
Ovarian, other
32 (40-49)
14 (60-69)
BRCA2a,c–f
HBOC
19 (20-29)
26%-91%
Ovarian, pancreatic, prostate, other
10 (40-49)
11 (60-69)
p53g–i
Li-Fraumeni syndrome
1.46 overall
56% to 45 yr
Soft-tissue sarcoma, osteosarcoma, brain tumors, adrenocortical carcinoma, leukemia, other
5.96 (15-29)
>90% to 70 yr
PTENj–l
Cowden syndrome
~2-4
25%-50%
Thyroid (follicular and rarely papillary) endometrial, genitourinary, other
Bannayan-Riley-Ruvalcaba syndrome
Proteus
Proteus-like syndrome
STK11m,n
Peutz-Jeghers syndrome
~15
54%
Small intestine, colorectal, uterine, testicular and ovarian sex chord tumors, other
CDH1o–q
Hereditary diffuse gastric carcinoma
~3.25
39%
Lobular breast, diffuse gastric, other
LOW PENETRANCE
ATM (heterozygote)r–t
Ataxia-telangiectasia in homozygotes
~3-4
—
Undefined in heterozygotes
CHK2 (CHEK2)u–x
Li-Fraumeni variant
2 (women)
—
Undefined
10 (men)
BRIP1y
Fanconi anemia in heterozygotes/compounds
2
—
Undefined in heterozygotes
PALB2z
None known
2.3
—
Undefined in heterozygotes
BARD1
None known
—
Undefined in heterozygotes
None known
4.3
—
Undefined in heterozygotes
XRCC2aa
None known
—
Genetics
Clinical Management
Cowden Syndrome
Clinical Features
Genetics
Risk Management Recommendations
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Genetic Factors: Hereditary Cancer Predisposition Syndromes