Figure 43-1. Cancer genetic counseling process.
Clinical genetic providers summarize the family history using a pedigree format. The family history can quickly be constructed, and by its very structure, biological relations are defined and patterns of cancer, and other key clinical characteristics, may be visualized. Three generation histories are typically obtained documenting individual family members, their children, and the family ethnic background. Recording extended relatives allows for the scope of at-risk family members to be recognized. The age at diagnosis, cancer type, as well as key exposure history, are solicited. When feasible, self-reported family histories are completed in advance of the genetics evaluation, allowing for family members to be contacted or medical records obtained. Limited research suggests that family history questionnaires completed in advance provide thorough family history information (J Genet Counsel 2009;18:366).
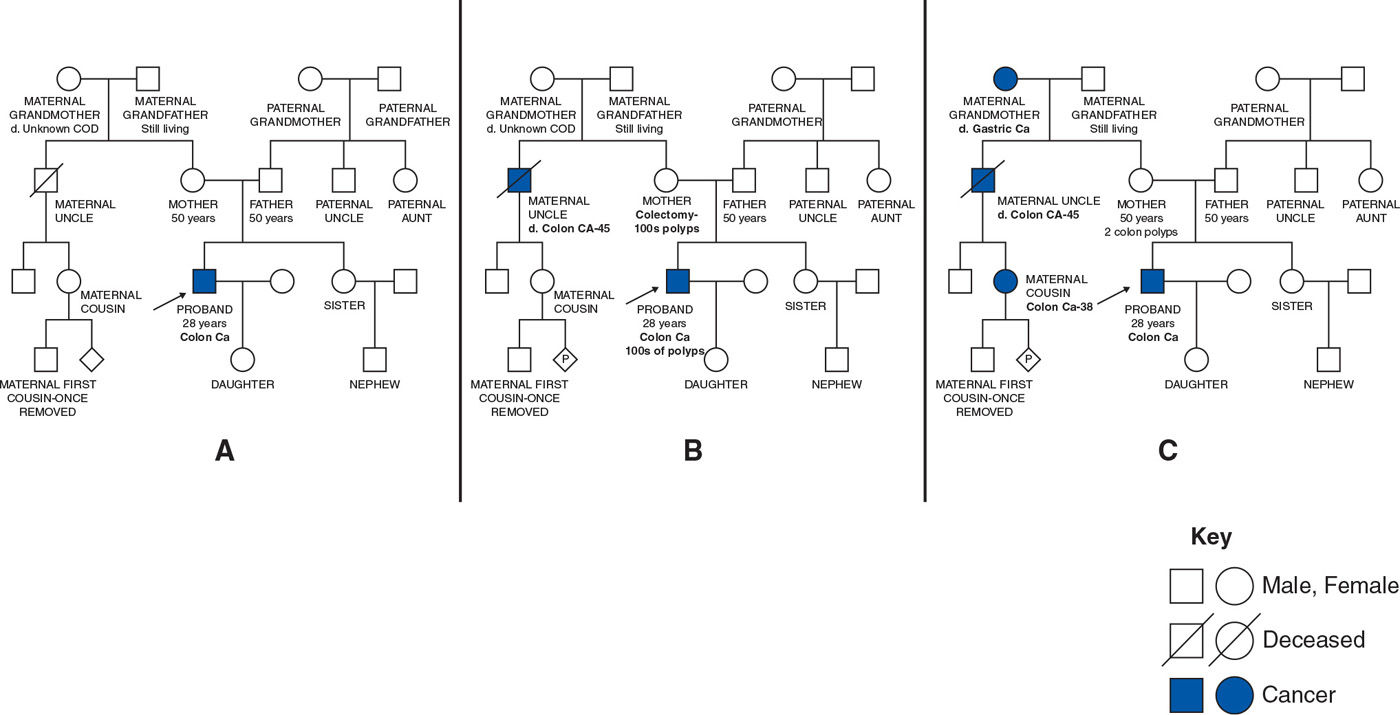
Consider a 28-year-old man diagnosed with colon cancer (Fig. 43-2). There is no reported family cancer history in the first example (Fig. 43-2A), and the genetics assessment is based solely on the age at diagnosis and tumor characteristics. Minimal changes to the reported family cancer history, as demonstrated in Figure 43-2B, leads to a diagnosis of familial polyposis based on the number of colonic polyps and dominant pattern of inheritance. Slightly different cancer reporting, as noted in Figure 43-2C, also demonstrates dominant inheritance but with a cancer pattern now diagnostic for the Lynch syndrome. Familial polyposis and Lynch syndrome are both hereditary colon cancer predisposition syndromes; however, they result from mutations in different genes, have distinct cancer risks, and consequently the medical recommendations differ. Visualization of the family history provides diagnostic and practical advantages.
There are several personal and family history features suggestive of hereditary disease (Table 43-1). To date, most, but not all, hereditary cancer syndromes are inherited following an autosomal dominant pattern of inheritance. Multiple affected individuals, who are closely related to one another, with more than one generation affected, are characteristics suggestive of any dominant disease. Young age at diagnosis, more than one primary cancer in a single individual, bilaterality of paired organs, the occurrence of rare tumor types, and unusual cancer presentations are key features. Distinct histologic features of specific cancers and specialized tumor genetic studies (e.g., microsatellite instability (MSI) testing) may provide important clues. It is the combination of features, not a single trait, that leads to the suspicion, or diagnosis, of inherited cancer predisposition. More specific clinical criteria, dependent on the predominant cancer in the family, have been developed (J Med Genet 2004;41:81). Independent evaluation of the maternal and paternal side of the family is necessary as the two histories are not additive.
Clinical assessment of the family cancer history may be hindered by the level of accuracy of reported cancer history, family structure, and the general age of the family. Overreporting the family cancer history may lead to an overestimation of risk with implementation of inappropriate medical interventions. In contrast, underreporting the family cancer history may result in lower cancer risk estimates with consequential underutilization of potentially beneficial surveillance and or prophylactic medical and surgical interventions.
Several studies have assessed the validity of cancer reporting, using both cancer and general populations, with evaluation of reporting for several different cancer types (J Med Genet 1999;36:309; J Natl Cancer Inst 2011;103:788; JAMA 2004;292:1480; Am J Prev Med 2003;24:190). Key findings include the following: (a) cancer reporting for first-degree relatives is more accurate than for second- or third-degree relatives; (b) cancer populations, who may be more motivated to seek family history information and demonstrate higher levels of reporting accuracy than general populations; (c) reporting accuracy varies by cancer site with breast and colon cancer consistently being correctly reported among first degree relatives; and (d) younger informants tend to have higher reporting accuracy (J Med Genet 1999;36:309; J Natl Cancer Inst 2011;103:788; JAMA 2004;292:1480; Am J Prev Med 2003;24:190).
Personal and Family History Features Suggestive of Hereditary Cancer |
• Multiple affected individuals who are closely related to one another
• Cancers occurring in multiple generations
• Clusters of the same type of cancer (e.g., brother, father, paternal uncle with prostate cancer)
• Young age at diagnosis (e.g., colon cancer diagnosed at 35 yr)
• Multiple primary cancers in a single individual (e.g., sarcoma and breast cancer)
• Bilaterality in paired organs or multifocal disease (e.g., bilateral renal clear cell cancer)
• Uncommon presentation (e.g., male breast cancer)
• Rare cancer types (e.g., retinoblastoma, paraganglioma)
• Unusual tumor histology (e.g., renal chromophobe histology)
• Tumor genetic studies designed to screen for specific cancer syndromes (e.g., MSI analysis)
• Presentation of unusual skin lesions (e.g., mucocutaneous hyperpigmentation of mouth and lips)
• Combination of cancers suggestive of a known cancer syndrome
• Ethnic background associated with higher incidence of specific gene mutations
To verify the reported cancer history, medical records and/or death certificates are collected. Pathology, medical, and surgical records are the most informative; however, these records are not always available given the limited time health-care facilities maintain paper records. Widespread implementation of electronic medical record systems will help to address this issue. Death certificates are inexpensive and readily available through a state’s vital statistics office and may confirm a family member’s cancer type and age at diagnosis.
Family structure may also impact the evaluation of the family’s cancer history. Consider a woman with a pancreatic neuroendocrine tumor. Each parent was an only child with no siblings. This woman has no biological aunts, uncles, or cousins. Simply put, there are a limited number of people or “data” to evaluate the family history. The relative “age” of a family may also lead to a biased assessment. The parents of the proband in Figure 43-2a are only 50 years of age, and nearly 10 years younger than the average age of diagnosis for many common cancer types. It is important to understand that the family’s cancer history may not have fully expressed itself yet when evaluating a young adult with cancer or a “young” family.
II. HEREDITARY CANCER PREDISPOSITION SYNDROMES. If the personal and family history features are consistent with hereditary disease, then the question is, what is the underlying cancer syndrome in the family?
The number of cancer predisposition syndromes described continues to grow (Table 43-2) (J Natl Cancer Inst Monogr 2008:1; Fam Cancer 2013;12:1). Overlapping clinical features (cancer risks) may necessitate the consideration of multiple diagnoses for a given family. Diagnostic guidelines have been established for many syndromes to assist in their clinical recognition (Cancer Res 1988;48:5358; Genetic/familial high-risk assessment: breast and ovarian. www.nccn.org, 2013; Gastroenterology 1999;116:1453).
Most of the cancer syndromes delineated to date are highly penetrant, dominantly inherited disorders. Penetrance is the frequency in which a gene or gene combination manifests itself in a gene carrier. Every person who carries a gene mutation in a disease with 100% penetrance will develop feature(s) of that disease, whereas only 30% of individuals will develop feature(s) in a syndrome with 30% penetrance. For cancer syndromes, high penetrance translates to high risk for certain cancer types. Expressivity is the term used to describe the clinical variability of a specific disease, and in the case of hereditary cancer is the spectrum of benign and malignant tumors associated with that cancer predisposition syndrome. Much of these data are currently derived from highly selected families, which may represent the extreme end of the disease spectrum. The penetrance and expressivity of a given syndrome will be used to determine screening and medical management.
As the genetic heterogeneity of inherited cancer predisposition is unraveled, more cancer syndromes will be defined, following varying patterns of inheritance, and with low-to-modest penetrance rates. Syndrome recognition will become increasingly more challenging leading to a greater reliance on genetic testing.
III. GENETIC TESTING. Genetic testing may encompass the analysis of gene(s), exomes, or even the entire genome. Genetic testing conducted to identify germline (inherited) gene mutations is typically performed on a blood, mouthwash, buccal swab, or banked DNA specimen. The purpose of germline genetic testing in the oncology setting is to identify the underlying genetic basis for the cancer predisposition in the family and subsequently use this information to guide treatment, follow-up, and make genetic testing available to at-risk family members.
In contrast, genetic analysis performed on a malignant tumor is primarily intended to characterize somatic (acquired) genetic aberrations for the purpose of identifying potential therapeutic targets. Tumor genetic testing may be used to screen a tumor for characteristics for a specific syndrome. For example, MSI analysis, performed on colon or endometrial malignancies, is used to screen for the Lynch syndrome (JAMA 2012;308:1555). Follow-up germline genetic testing is absolutely necessary to distinguish if the positive tumor screen resulted from a germline mutation or somatic events.
Some Hereditary Cancer Predisposition Syndromes |
Cancer syndrome gene(s) | Core clinical features | Primary medical recommendations beyond population-based cancer screening guidelines |
Familial adenomatous polyposis APC | High risk for 100s–1,000s of colon polyps. Without total colectomy, colon cancer will eventually develop Increased risk for small bowel, thyroid, hepatoblastoma, pancreas cancer Desmoid tumors (10%–20%), congenital hypertrophy of retinal pigmentation (CHRPE), gastric polyps, osteomas | • Consideration of screening for hepatoblastoma with liver ultrasound and serum alpha-fetoprotein concentrations up to 5 yr of age • Colonoscopy screening beginning by 10 yr of age Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|


