Genetic predisposition accounts for only a fraction of breast cancers, but genetic testing, in the appropriate context, is the most powerful tool available for quantitative risk stratification. The information provided by genetic testing contributes directly to risk management decisions and, frequently, to decisions about cancer treatment. BRCA1 and BRCA2 are still responsible for the majority of hereditary breast cancer cases, but the list of rare genes is increasing rapidly. The commercialization of massive parallel sequencing tests (i.e., next generation sequencing) has revolutionized genetic testing. Panel testing is becoming more common and patients are presenting more frequently with rare mutations in genes like CHEK2, PALB2, and ATM.
The introduction of paradigm-shifting technology is often associated with a period of controversy and uncertainty as the technology finds its place. This is true for next generation sequencing as well. Little data is available to accurately estimate cancer risks for many of the new genes and a high rate of uninterpretable results (i.e., variants of uncertain clinical significance) is expected. These issues will be resolved over time. The value of professional genetic counselors cannot be overstated in this period of uncertainty. This chapter provides an overview of recent advances in genetic testing as well as information about the new syndromes clinicians are encountering.
Lifetime breast cancer risk is only 13% for identical twins of breast cancer patients as compared to 9% for dizygotic twins.1 Similar twin studies estimate that 12% to 30% of breast cancer is primarily genetic in origin2,3 and modeling studies implicate autosomal dominant inheritance of single genes as the most important mechanism.4,5 These data suggest that environment (e.g., reproductive factors) accounts for a greater number of breast cancers than genetics. Despite enormous advances in gene sequencing, it is estimated that only 35% of apparently familial breast cancer is accounted for by known mutations (Fig. 68-1).6 The balance is likely accounted for by very rare moderately penetrant single gene mutations or by inheritance of combinations of low-penetrance mutations (i.e., polygenetic mechanism). Genome-wide association studies have convincingly identified 21 low-penetrance mutations or polymorphisms with relative risks in the range of 1.1 to 1.3.7–12 Increasing numbers of mutations in an individual is associated with increasing breast cancer risk with the greatest risk conferred by the rarer mutations.13
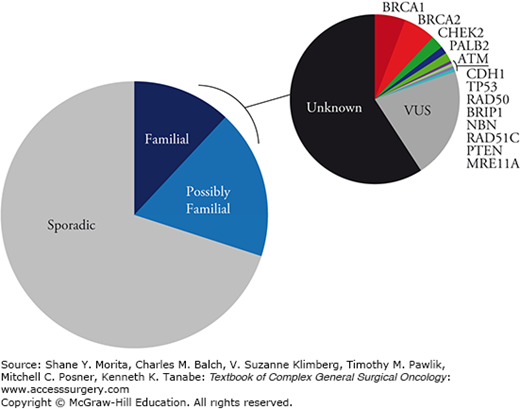
Mutations in some genes, such as BRCA1 or TP53, are classified as highly penetrant; that is, most individuals inheriting the mutation will develop cancer. Nevertheless, penetrance can vary greatly by family for any of the genes. This is likely due to the pattern of variants in other genes unique to each family. This is known as gene–gene interaction. There are many convincing examples of gene–gene interaction affecting the penetrance of known breast cancer predisposition genes.14–19 Careful evaluation of the three-generation pedigree can often provide some sense of the penetrance of a given mutation in a specific family. One could also postulate that certain environmental factors present within a family group could affect penetrance; however, convincing examples of gene–environment interactions are more difficult to come by.20
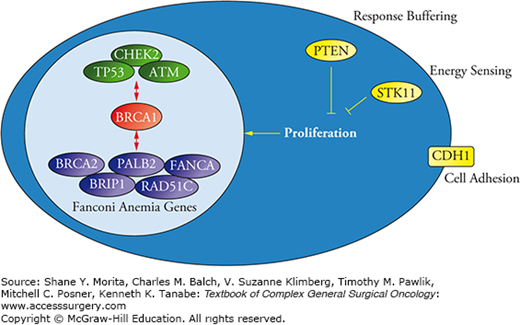
Most of the breast cancer susceptibility genes function in cell cycle control and/or DNA repair. Many are Fanconi anemia genes (Fig. 68-2). Recall that Fanconi anemia is an autosomal recessive (and occasionally X-linked) syndrome characterized by chromosomal instability, bone marrow failure, and a high risk for acute myelogenous leukemia. There are 16 Fanconi genes and the syndrome results when an individual inherits a mutated gene from both the mother and the father. Five Fanconi genes have been linked to increased breast cancer risk in the heterozygous state: FANCD1/BRCA2, FANCJ/BACH1/BRIP1, FANCN/PALB2, FANCO/RAD51C, and FANCA.21 Each of these is known to interact with BRCA1. Non-Fanconi DNA repair genes linked to breast cancer predisposition also interact directly or indirectly with BRCA1 and include TP53, ATM, and CHEK2.22 Other breast cancer predisposition genes that are not, apparently, integral to DNA repair include PTEN, a protein that buffers proliferation signals coming from outside the cell; STK11, a protein that blocks proliferation when environmental conditions are not optimal for cell division; and CDH1, a protein that normally inhibits proliferation when a cell is in contact with other cells.
Genetic tests are performed on germline DNA that is obtained from white blood cells or from oral squamous mucosal cells. A genetic test may be designed to assess a single nucleotide in one or a few genes (e.g., single site tests or the Ashkenazi BRCA panel) or may derive the nucleotide sequence for large portions of one, a few, or many genes. Classic sequencing tests in use for the last few decades were designed to assess portions of the promoter, some or all of the exons, and parts of the introns that may be involved in mRNA splicing. While highly sensitive for identifying deleterious single nucleotide changes, this approach is blind to deletions, insertions, or duplications of longer nucleotide sequences which may also be deleterious. In the case of BRCA gene testing, additional testing for large rearrangements is now routinely performed at the time of initial gene sequencing; however, clinicians should be aware that tests performed prior to 2012 may not have included this testing. This should be considered when interpreting a negative genetic test.
While classic DNA sequencing (Sanger sequencing) derives nucleotide sequences from one PCR-amplified section of DNA at a time based on the last (terminating) nucleotide added to variable length fragments of that DNA, Next Generation Sequencing (NGS) can fragment and amplify DNA from the entire genome and then simultaneously derive the sequence of millions of short fragments based on the order that complementary nucleotides are added to those fragments. This, in effect, permits simultaneous sequencing of the entire genome. The process generates enormous volumes of data that must then be processed to produce useful information. This is not trivial as each test will identify millions of variants, only a few or none of which may be clinically relevant. Initial processing uses statistical approaches to remove noise (i.e., sequence calls that do not show a sufficiently high level of reproducibility) and then various algorithms, based on public and proprietary data, are used to classify the remaining variants as deleterious, uncertain clinical significance, suspected benign, or definitely benign. Deleterious mutations are typically confirmed using focused Sanger sequencing, and specific genes may be further interrogated by any number of tests to identify or confirm large rearrangements.
Several companies are now offering next generation sequencing tests that assess multiple genes grouped into panels that include all of the genes known to be implicated in a specific disease (e.g., breast cancer). The consumer must now choose between these tests, but there is very little comparative data available to guide this choice. At the time of this writing, major technical differences in the tests that might impact sensitivity and specificity are not apparent and available data suggest that NGS tests are reliable.23 The tests vary widely in cost, but this may not be the best criterion for choosing a test. Some companies attempt to differentiate themselves based on the additional testing they do to confirm deleterious point mutations and large rearrangements. Another factor to consider is the frequency of variants of uncertain clinical significance and the approach the company is using to get these variants reclassified as either deleterious or definitely benign. Companies that have acquired considerable experience with specific populations of patients and that employ reliable systematic approaches for reclassifying VUS calls are likely to most consistently provide clinically actionable results.
The American Civil Liberties Union and the Public Patent Foundation filed a law suit against Myriad Genetics, Inc, previously the sole provider of BRCA gene testing in the United States, on May 12, 2009, challenging their BRCA1 and BRCA2 gene patents. On June 13, 2013 the Supreme Court ruled that isolated genomic DNA is not eligible for patent protection. Within a week several other companies began offering BRCA gene testing, but some of these companies were sued for patent infringement. Many of these lawsuits remain unsettled at the time of this writing.
A genetic test result may be the most definitive factor available for individual breast cancer risk estimation. The first task after receiving the result is to update the cancer risk projections in order to develop a rational management plan. The results of a genetic test may be classified as deleterious, variant of uncertain clinical significance, variant suspected benign, or normal (Table 68-1). A deleterious mutation in a high-penetrance gene, such as BRCA1 or BRCA2, is easy to interpret. When this result is obtained it is straightforward to work through a family using single site testing to accurately classify each family member as high risk or not high risk. For this reason, it is always desirable to first test the individual in a family that is most likely to carry a deleterious mutation. This is generally someone who has already been diagnosed with the relevant cancer, usually at an early age. However, the advent of next generation panel testing introduced additional difficulties in interpretation.
Interpreting Genetic Test Results
| Test Result | Interpretation Challenge |
|---|---|
| Deleterious | Penetrance (how much cancer risk)
|
| Variant uncertain clinical significance |
|
| Variant suspected benign |
|
| Definitely benign/Negative |
|
Though the panels include genes definitely associated with high cancer risk (i.e., high-penetrance genes), such as BRCA1, BRCA2, TP53, and PTEN, they also include genes conferring only modest increased risk such as CDH1, ATM, and CHEK2, as well as genes thought to be associated with only very small increases in breast cancer risk such as MUTYH or NBN. There is a paucity of epidemiological data available for many of the genes included in the panels, making risk estimation a moving target. For example, PALB2 was initially classified as a modest-penetrance gene, but apparent high-penetrance families are now being identified, suggesting that it may be similar to BRCA2 in some families.24 This highlights the importance of going back to the three-generation pedigree when interpreting a genetic test result in order to get a sense of the penetrance in a particular family. This approach is supported by observations in BRCA2 families suggesting family-specific penetrance patterns.25
Early experience with the Ambry Genetics BreastNext panel, which included 14 genes at that time (not BRCA1 or BRCA2), recorded a VUS rate of 33%.26 An update that included 400 patients did not report an overall VUS rate, but did note that the VUS rate was highest for ATM, BRIP1, and BARD1 and lowest for STK11, TP53, and CDH1.27 A recent report that included 31 BreastNext tests reported a VUS or variant suspected benign rate of 23%.28 In these early years of NextGen panel testing, patients and providers need to be aware that the test may yield an inconclusive result one time out of four. This is a temporary situation, however. In the initial years that Myriad Genetics Inc. offered BRCA1 and BRCA2 gene sequencing, the VUS identification rate was 7% to 15%.29,30 By systematically addressing these variants through more extensive testing in certain families and various informatics tools, this rate has been reduced to 2.9%.31 VUS rates are somewhat higher for non-Caucasian populations, but have declined from 22–46%32,33 to 2.6–7.8%31 in recent years. A recent review describes current approaches for classifying mutations, and provides online resources for interpreting specific VUS results.34 When a genetic test returns a VUS result, it must be acknowledged that the test has not been helpful in classifying risk. Ideally, the patient’s result and contact information need to be maintained so that they can be contacted when and if the variant is reclassified.
Commercially available panel tests focus on collections of genes that are known or suspected to be involved in breast cancer predisposition. Many of the genes were only recently recognized as putative predisposition genes and, for some, it is not certain that they really are. Some of the best evidence for a breast cancer link comes from segregation studies in large families where the mutation is seen to occur in family members diagnosed with breast cancer, but not in unaffected family members. Incomplete segregation has been observed for some of the new genes,6 suggesting that they may not actually be responsible for the breast cancers observed in a family or that they require coexisting mutations in other genes to exert an effect. An example is RAD51C which appeared to be strongly associated with breast cancer in initial studies35 but not later studies.36 The challenge, then, is to accurately translate a genetic test result into a quantitative cancer risk profile. For now, we are counting on the test provider to do the bulk of the background work to accomplish this, but the value of a three-generation pedigree in the hands of an experienced genetic counselor cannot be overstated. Pretest counseling is more important than ever for framing the discussions that might ensue after the test result is received, but with the introduction of multigene tests the focus is shifting away from thinking about which genes to test and toward interpreting the test results.
In addition, wider use of whole-genome sequencing tests in clinical practice has exposed new ethical challenges surrounding the disclosure of incidental findings.37 An example is the identification of a deleterious BRCA2 mutation in a 5-year-old boy undergoing exome sequencing for evaluation of developmental delay. Disclosure of this result to the patient and his family could violate autonomy (i.e., the right not to know), but not disclosing it violates a fiduciary duty to prevent harm by warning. The American College of Medical Genetics and Genomics has published thoughtful guidance on these issues.38,39 Pretest counseling should establish the patient’s preference for receiving information on incidental findings, and only monoallelic high-penetrance mutations should be reported.
Mutations in BRCA1 and BRCA2 are still the most frequently identified germline mutations in hereditary breast cancer. Other deleterious mutations identified through panel testing, in descending order of frequency, include CHEK2, PALB2, ATM, CDH1, TP53, RAD51C, PTEN, and STK11. Table 68-2 provides a brief update on these syndromes.
Mutation Frequency, Breast Cancer Risk, and Clinical Features of Some of the Recognized Syndromes
| Gene | Clinical Mutation Positivity Ratea | Lifetime Breast Cancer Risk | Clinical Notes |
|---|---|---|---|
| BRCA1 | 5–10% | 65–81% | Basal-type triple negative breast cancer; ovarian cancer |
| BRCA2 | 5–10% | 45–85% | Hormone-sensitive breast cancer; male breast cancer; melanoma; pancreas cancer |
| CHEK2 | 0–3% | 20–44% | Hormone-sensitive breast cancer |
| PALB2 | 1–4% | 25–91% | Hormone-sensitive breast cancer; male breast cancer; pancreatic cancer |
| ATM | 1–3% | 15–60% | Certain mutations increase risk, others do not; difficult to interpret results |
| CDH1 | <1% | 39–52% | Hereditary diffuse gastric cancer; infiltrating lobular carcinoma |
| TP53 | <1% | >50% | Very early onset high-grade, hormone-sensitive, and HER2-positive breast cancer |
| RAD51C | <1% | 10–20% | Ovarian cancer families |
| PTEN | <1% | 77–85% | Cowden syndrome; macrocephaly, extensive benign breast disease, thyroid and endometrial cancer |
| STK11 | <1% | 24–32% | Peutz–Jegher syndrome; mucocutaneous pigmentation; hamartomatous polyps; early onset breast cancer; pancreas and GI cancer |
Lifetime breast cancer risk ranges from 60% to 81% for BRCA1 mutation carriers and 45% to 85% for BRCA2 carriers.40–43 The mean age at diagnosis of breast cancer is about 44 years for BRCA1 mutation carriers and 47 years for BRCA2 carriers, but age at onset varies by family, particularly for BRCA2 families.44–46 Both syndromes are associated with increased ovarian cancer risk, but the risk is higher for BRCA1. Both increase the risk of male breast cancer, but the risk is higher for BRCA2. BRCA2 is also associated with increased risk for melanoma, pancreas and other cancers, but the risk is not high enough to warrant systematic screening.
Checkpoint kinase 2 (CHEK2) cooperates with ATM, p53, and BRCA1 to facilitate accurate chromosomal separation during cell division.47–49 An inactivating mutation in the kinase domain, CHEK2 1100delC, was reported to be associated with increased breast cancer risk in 2002.50 Most subsequent work has focused on this one mutation, but comprehensive analysis of the gene has identified other deleterious mutations.51,52 CHEK2 mutations are identified in 0% to 3% of breast cancer patients depending on geographic region and ethnicity.51,53–59 CHEK2 is a modest penetrance breast cancer susceptibility gene with relative risks in the 2.3 to 3.7 range depending on how the families are identified.60 There is evidence for variable penetrance between families so a review of the pedigree is particularly valuable for estimating breast cancer risk when a CHEK2 mutation is found. One study that included 277 mutation-positive probands estimated lifetime breast cancer risk at 20% if there was no family history of breast cancer, 28% for one affected second-degree relative, 34% for one affected first-degree relative, and 44% for both first- and second-degree relatives.57 It is likely that CHEK2 mutations increase the risk of contralateral breast cancer but data are inconsistent.55,61,62 Likewise, CHEK2 mutation may increase male breast cancer risk,63,64 but this is not consistently observed.65 Seventy percent to 80% of CHEK2 breast cancers are estrogen receptor positive66–68 and there is some evidence that mutations are associated with increased breast cancer-specific mortality among these women.55 Unaffected CHEK2 mutation carriers likely reach risk thresholds sufficient to consider enhanced surveillance with MRI, and because the majority of subsequent breast cancers are estrogen receptor positive, chemoprevention can be considered. Newly diagnosed breast cancer patients with CHEK2 mutations should be offered the full range of treatment options available to sporadic breast cancer patients.
There is some evidence that CHEK2 mutation carriers may be at increased risk for cancers other than breast. A large Polish study found that truncating mutations were also associated with an increased risk for common cancers such as thyroid, kidney, colorectal, and prostate,69 though a multicenter study confined to the CHEK2 1100delC mutation in Northern and Western Europeans did not find an increased risk for any cancer other than breast.70 Other studies and meta-analyses have suggested that CHEK2 mutations modestly increase the risk for melanoma,71 nonpolyposis colorectal cancer,72 prostate cancer,73,74 and bladder cancer.74 Six percent of 65 Li–Fraumeni-like families negative for p53 mutation were found to carry the CHEK2 1100delC mutation,75 and there is at least one CHEK2 family manifesting the full spectrum of Li–Fraumeni cancers.76 This is unusual, however, as the cancer risks tend to be organ-specific within families (e.g., colon in one family, breast in another).77 At any rate, the risk for other cancers is generally quite modest and no specific screening or management would be justified unless the pedigree suggested otherwise.
Partner and localizer of BRCA2 (PALB2) is a Fanconia anemia gene (FANCN)21 that gets BRCA2 to BRCA178 for accurate DNA repair.79 PALB2 mutations are found in 1% to 4% of BRCA1/2-negative familial breast cancer cases80–84 depending on the geographical region, with most reports clustering around 1% or less. PALB2 has been associated with pancreatic cancer risk85 and with male breast cancer.86,87 An initial report suggested that PALB2 mutations were absent in Ashkenazi Jewish populations,86 but some mutations have since been identified.88 PALB2 was initially classified as a moderate penetrance breast cancer susceptibility gene with relative risks in the 2.3 to 6 range,80,86,89 but high-penetrance families have been described (Fig. 68-3)90 and one study of five families estimated a 91% risk by age 70 with a median age at diagnosis of 42.24 Like BRCA2, the majority of PALB2-associated breast cancers are estrogen receptor positive.91 Clinically, the breasts of PALB2 mutation carriers can be evaluated and managed the same as BRCA2 carriers. Available data do not suggest elevated ovarian cancer risk.
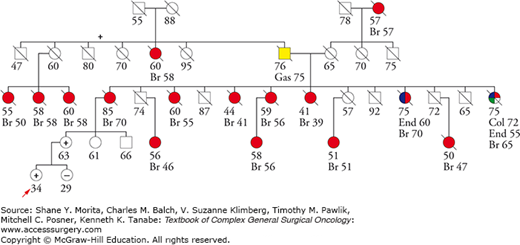
Ataxia-telangiectasia (A-T) is an inherited autosomal recessive neurodegenerative disease associated with an increased risk for leukemia and lymphoma. Increased breast cancer risk has not been reported, but these patients often succumb to the disease in early adulthood. Epidemiological studies have suggested that mothers of affected children (i.e., obligate heterozygous carriers) have an increased risk for breast cancer with relative risks in the 5 to 8 range.92,93 This raises the possibility that the 1% of the population who carry heterozygous ATM mutations94,95 may also be at increased risk.
The ataxia-telangiectasia mutated (ATM) gene is a prime breast cancer susceptibility gene candidate because of the way it cooperates with CHEK2 and BRCA1 for precise DNA double strand break repair.22,96 Many ATM mutations have been linked to breast cancer risk in initial studies, but not confirmed in larger studies or meta-analyses.97–102 However, it is likely that some rare monoallelic ATM mutations modestly increase breast cancer risk with relative risks in the 1.5 to 3 range.103–106 These are usually rare mutations in evolutionary conserved regions or specific functional regions and are often recognized as classic (i.e., A-T causing) mutations. Rarely, segregation analysis or case-control data has suggested that certain mutations in certain families may be associated with up to 40% to 60% breast cancer risk by age 80.107,108
Though homozygous A-T patients should definitely avoid radiation if possible,109 there is no convincing data to suggest that heterozygous ATM mutation carriers experience significantly increased toxicity from radiation.110,111 Though one study found that breast conservation with whole-breast radiation was associated with increased contralateral breast cancer risk for certain heterozygous ATM carriers (relative risk about 5),112 other studies have not observed an increased incidence of contralateral breast cancer.113,114
Related posts:
 Defining the Specialty of Surgical Oncology
Defining the Specialty of Surgical Oncology
 Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Palliation Of Incurable Gastric Cancer
Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Palliation Of Incurable Gastric Cancer
 Cystic Lesions of the Liver and Biliary Tract
Cystic Lesions of the Liver and Biliary Tract
 Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


