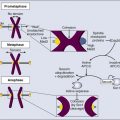
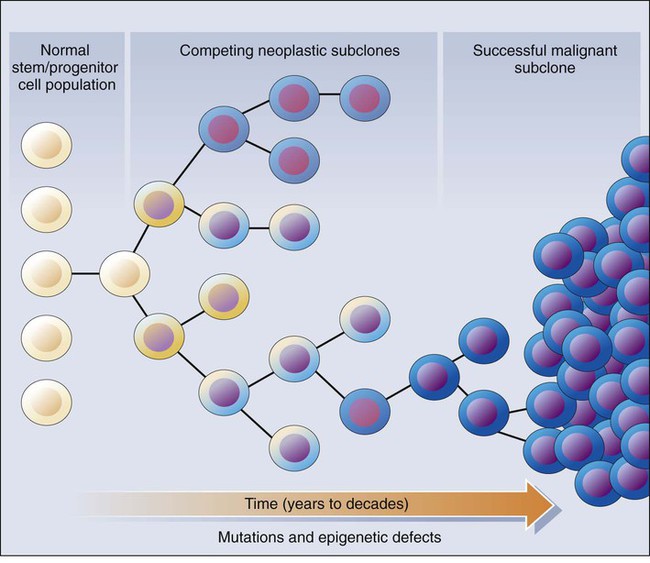
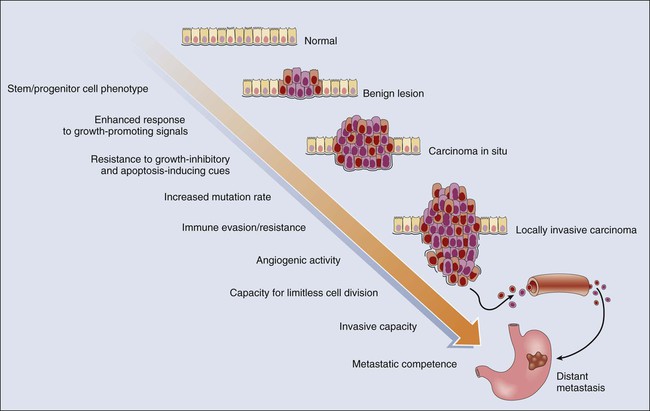
• A root cause of cancer is the accumulation of genetic and epigenetic defects in key cellular pathways regulating proliferation, differentiation, and death. The defects in cancer cells are of two types: gain-of-function alterations affecting oncogenes and loss-of-function alterations affecting tumor suppressor genes. Regardless of whether the defects are genetic or epigenetic in nature, a common net consequence is dysregulation of gene expression in cancer cells. • Clinical and pathological studies indicate that many cancers arise from preexisting benign lesions, and numerous cooperating genetic and epigenetic defects affecting multiple independent signaling pathways are likely needed for development of most clinically recognizable cancers. • A process termed clonal selection has a key role in determining the particular constellation of genetic and epigenetic defects present in a cancer cell. Clonal selection is essentially an evolutionary process that promotes outgrowth of precancerous and cancerous cells carrying those mutations and gene expression changes that confer the most potent proliferative and survival properties upon the cancer cells in a given context. • Although a sizeable and diverse array of mutations and gene expression changes have been implicated in cancer pathogenesis, the defects appear to affect a more limited number of conserved signaling pathways or networks. A relatively small collection of oncogenes and tumor suppressor genes is recurrently deranged in cancer cells of various types and includes the RAS, PIK3CA, EGFR, RAF, β-catenin, and MYC oncogene proteins and the p53, p16Ink4a, ARF, RB1, PTEN, APC, and NF1 tumor suppressor proteins. The proteins that are recurrently targeted by mutations in cancer likely represent particularly critical hubs in the cell’s regulatory circuitry. • Although cancer represents a very heterogeneous collection of diseases, the development of all cancers, regardless of type, appears to be critically dependent on the acquisition of certain traits that allow the cancer cells to grow in an unchecked fashion in their tissue of origin and to grow as metastatic lesions in distant sites in the body. Signature traits that are likely to be inherent in the majority, if not all, cancers include the following: (1) an increased tendency to manifest a stem cell or progenitor-like phenotype; (2) an enhanced response to growth-promoting signals; (3) a relative resistance to growth inhibitory cues; (4) an increased mutation rate to allow for the rapid generation of new variant daughter cells; (5) the ability to attract and support a new blood supply (angiogenesis); (6) the capacity to minimize an immune response and/or evade destruction by immune effector cells; (7) the capacity for essentially limitless cell division; (8) a failure to respect tissue boundaries, allowing for invasion into adjacent tissues and organs, as well as blood vessels and lymphatics; and (9) the ability to grow in organ sites with microenvironments that are markedly different from the one where the cancer cells arose. • Certain gene defects in cancer cells may contribute to a few or perhaps even only one of the signature traits. However, many of the gene defects and expression changes might have been selected for in large part because they exert pleiotropic effects on the cancer cell phenotype. • Despite the fact that some gene defects may arise early in the development of certain cancer types, advanced cancer cells might still be critically dependent on the “early gene defects” for continued growth and survival. Such findings imply that agents that specifically target key signaling pathways and proteins could have utility in advanced cancers even if the signaling pathway defect arose very early in cancer development. • Future studies will further clarify the role of the diverse array of genetic and epigenetic defects in cancer phenotypes, allowing more definitive and more specific strategies for cancer detection and diagnosis and therapeutic targeting of cancer cells. • Genomic characterization of organ site cancer—breast cancer, for example—identifies a number of subtypes with different prognoses and therapeutic relevance. Given this background, it would appear that compelling evidence exists that cancers arise from accumulated defects affecting multiple genes and pathways, and precancerous (benign) precursor lesions harbor fewer of the key gene defects. One question, therefore, is how many rate-limiting defects or “hits” are required for cancer development. Although a definitive answer to this question cannot be given at this point, some estimates can be offered. Most common cancers show dramatically increased incidence with increasing age. On the basis of analysis of the age-specific incidence of a number of common cancers and some straightforward assumptions about the rate of mutations and the size of the target cell population, it was argued as early as the mid 1950s that most common epithelial cancers arise as the result of four to seven rate-limiting events.1,2 It was inferred that these rate-limiting events might represent mutational events. Moreover, benign lesions were inferred to arise as the result of fewer gene defects, consistent with the fact that recognizable benign lesions that are often found show an age-incidence distribution that was shifted roughly one to two decades earlier in life than cancers arising in the corresponding organ or tissue sites. Nevertheless, confounding the use of age-incidence data to model the number of rate-limiting mutations were questions about certain key biological assumptions underlying the multihit models. Given the attendant uncertainties with estimates of rate-limiting mutation numbers derived largely or solely from age-incidence data and the practical difficulties in defining the nature and significance of all inherited and somatic gene defects in cancer, a definitive answer to the number of rate-determining defects in a particular cancer type has not yet been obtained. For instance, even one of the early comprehensive molecular analyses of breast and colon cancer indicated that dozens to hundreds of somatic mutations were often present in a given cancer,3 without even considering the even far greater number of epigenetic and gene expression changes present in the cancer cells relative to adjacent normal cells. Study of the genetic alterations of pediatric cancers that occur much earlier and are perhaps less complex in terms of their genetic signature may offer insights into key cancer target genes (see Chapter 95 on pediatric solid tumors). These molecular findings in benign and malignant tumors are essentially consistent with a model initially proposed by Foulds4 and subsequently advanced by Nowell5 (Fig. 13-1). In brief, the clonal evolution model predicts that cancers arise as the result of successive expansions of clonally related cell populations. The successive expansions are driven by the gradual or punctuated acquisition of mutations and gene expression changes that endow a particular cell and its progeny with a selective advantage over cells that do not harbor the gene defects. In essence, clonal selection is an evolutionary process that allows the outgrowth of precancerous and cancerous cells that carry mutations and gene expression changes that confer the most potent proliferative and survival properties on the cancer cells. It is important to note that the specific constellation of genetic and epigenetic changes that are present in precancerous and cancerous cells is context-dependent and certainly varies considerably from one cancer type to another and even most certainly varies to a significant degree among patients whose cancers display quite similar clinical and histopathological features. The basis for the context-dependent relationship of the changes that confer a selective growth advantage in a particular cancer may reflect physiological differences in organ site and the tissue microenvironment within the organ site, the identities of the preceding somatic gene defects in the precancerous or cancerous clone, and even the constitutional sequence variations and particular gene expression patterns that are present in nonneoplastic tissues of a given patient. This issue of context-dependent effects of gene defects that promote clonal selection in neoplastic cells will be addressed further in the following sections. Recent comprehensive sequence-based analyses of cancer cell populations from individual patients in which the tumor cells under study were distinct from one another in location and/or time has begun to reveal that quite dramatic intratumoral genetic heterogeneity may be a “rule” in cancer, rather than an exception.6 Certain, perhaps key, initiating genetic lesions might be shared among all neoplastic clones, but geographically distinct regions of large primary tumors may have very distinct mutation profiles from those in other portions of the primary tumor, and the metastatic cell populations may have considerable mutational divergence from the nonmetastatic cells. As such, it seems that quite extensively branched evolutionary growth may be an important feature in both primary tumors and metastatic lesions, with multiple competing clonal populations evolving through divergent and convergent mutational mechanisms.6 This more recent view of the potentially quite extensive intratumoral genetic heterogeneity in any given cancer and the contributions of intratumoral heterogeneity to tumor progression contrasts with some earlier views. Prior to the recent studies, it was suspected that the cell populations in many primary cancers might be more homogeneous, where somatic mutations were accumulated in a more stepwise fashion as a result of multiple sequential clonal sweeps of each variant cell populations in the primary cancer, with metastases perhaps most often arising from the clonally dominant cell population in the primary tumor. It is important to note that clonal somatic mutations are often presumed to have a causal role in promoting further tumor outgrowth or progression because somatic mutations can become clonal (i.e., present in all neoplastic cells) by only a limited number of mechanisms. For instance, the genetic alteration itself could have been selected for because it provided the neoplastic cell with a growth advantage, allowing it to become the predominant cell type in the tumor (clonal expansion). Genes with critical roles in promoting clonal outgrowth in a given cancer have been termed drivers.7 Alternatively, a somatic mutation, when detected, might have arisen essentially coincident with another, perhaps undetected, alteration that was the crucial change underlying clonal outgrowth. Somatic mutations of this latter type have been termed passengers.7 Genes that are mutant in a significant fraction of cancers and for which other lines of evidence link them to the cancer process can more readily be classified as drivers. However, on the basis of early data from some large-scale sequencing analyses that reveal large numbers of distinct genes that are each mutated in only a minority of cancers of a given type,3,8–10 sorting out drivers and passengers might not be entirely straightforward, based solely on sequencing data, but will likely require a significant body of functional studies and data. Statistical arguments may fall short, at least before large volumes of sequencing are obtained for the alterations that are private or rare, yet still biologically important when present in a given patient’s cancer. Cancer represents a highly heterogeneous collection of diseases. Each cancer type has distinct biological and clinical features and a variable prognosis. Even cancers that arise at a single organ site, such as the ovary, kidney, or lung, represent a hodgepodge of different diseases at the molecular level. Morphologic features often allow the particular cancer types to be distinguished to some degree from one another. Yet even for patients whose cancers have essentially identical gross and microscopic appearances and very similar clinical manifestations, there may be vast differences in outcome. In spite of this complexity, the development of all cancers, regardless of type, is likely to be critically dependent on the acquisition of certain phenotypic features that allow the cancer cells not only to grow in an unchecked fashion in their tissue of origin but also to gain the ability to disseminate into surrounding tissues and organs, lymphatics, and the bloodstream and ultimately to grow as metastatic lesions in distant sites in the body.11 As is indicated in Figure 13-2, among the signature traits that are likely to be expressed in the majority, if not all, of cancers are the following: (1) an increased tendency to manifest a stem cell or progenitor-like phenotype; (2) an enhanced response to growth-promoting signals; (3) a relative resistance to growth-inhibiting cues; (4) an increased mutation rate to allow for the rapid generation of new variant daughter cells; (5) the ability to attract and support a new blood supply (angiogenesis); (6) the capacity to minimize an immune response and/or evade destruction by immune effector cells; (7) the capacity for essentially limitless cell division; (8) a failure to respect tissue boundaries, allowing for invasion into adjacent tissues and organs as well as blood vessels and lymphatics; and (9) the ability to grow in organ sites with microenvironments markedly different from the one where the cancer cells arose. The development of some traits is likely to be associated with certain stages of tumorigenesis (see Fig. 13-2), but acquisition of signature traits in cancers is more likely to show a preferred order than an invariant order. Furthermore, many of the signature traits of cancer cells represent complex biological capabilities (e.g., angiogenic activity, immune evasion/resistance, and metastatic competence). Therefore it is likely that substantial changes in many signaling pathways are needed for the cancer cell to manifest these traits. As discussed previously, in genetic terms, oncogenic alleles have gain-of-function mutations. Oncogenic variant alleles that are present in cancer are generated from the normal counterpart proto-oncogenes by various mutational mechanisms, including point or localized mutations, gross chromosomal rearrangements, or gene amplification. Some representative oncogene mutations in human cancer are summarized in Table 13-1. From a brief review of the data in Table 13-1, some generalizations can be offered. First, the mutations affect proteins functioning in various compartments of the cell, including growth factor receptors, cytoplasmic signal transducers, and nuclear proteins, such as transcription factors. Second, although some oncogene mutations may be unique to cancers of a particular type, such as the specific chromosomal translocations and resultant fusion proteins that are seen in cancers of hematopoietic origin (e.g., the BCR-ABL translocation that is seen in chronic myelogenous leukemia and a subset of acute lymphoid leukemias and the PML-RARα translocation that is seen in acute promyelocytic leukemia), other mutations, such as those affecting the KRAS, β-catenin, and c-MYC genes, are found in a broad spectrum of different cancer types. Third, oncogene mutations in cancer are nearly always somatic, because only a very limited number of germline mutations in proto-oncogenes have been linked to cancer predisposition thus far. Fourth, some proto-oncogenes, such as KRAS or BCL2, are somatically altered in cancer by a single mutational mechanism, namely, point mutations in the KRAS gene and chromosomal translocations affecting the BCL2 gene. In contrast, other proto-oncogenes, such as c-MYC, may be activated by more than one mechanism in cancer, including chromosomal translocation and gene amplification. Both mutational mechanisms lead to increased levels of c-MYC transcripts and protein. In fact, specific missense mutations at threonine 58 of the c-MYC gene in some lymphomas may further enhance c-MYC protein levels by abrogating a phosphorylation-ubiquitination mechanism targeting c-MYC for proteosomal degradation.12 Of course, enhanced c-MYC protein levels can result from alterations in c-MYC–specific microRNAs, methylation, and other noncoding regulatory elements functioning to regulate c-MYC transcription and translation. Table 13-1 Selected Oncogene Mutations in Cancer
Genetic and Epigenetic Alterations in Cancer
Cancers Arise From the Accumulation of Multiple Gene Defects
Clonal Selection and Evolution in Cancer
Contribution of Gene Defects to the Signature Traits of Cancer Cells
Genetic and Epigenetic Defects in Cancer Alter Signaling Pathways and Regulators of Transcription, Chromatin, and Genomic Integrity
Recurrent Mutational Targets in Cancer
Gene
Activation Mechanism
Protein Properties
Tumor Types
KRAS
Point mutation
Signal transducer
Pancreatic, colorectal, lung (adeno), endometrial, other carcinomas
NRAS
Point mutation
Signal transducer
Myeloid leukemia, colorectal cancer
HRAS
Point mutation
Signal transducer
Bladder carcinoma
EGFR (ERBB)
Amplification, mutation
Growth factor (EGF) receptor
Gliomas, lung (non–small cell) carcinoma
NEU (HER2/ERBB2)
Amplification
Growth factor receptor
Breast, ovarian, gastric, other carcinomas
c-MYC
Chromosome translocation
Transcription factor
Burkitt lymphomas
Amplification
Small cell lung carcinoma (SCLC); other carcinomas; glioblastoma
MYCN
Amplification
Transcription factor
Neuroblastoma, SCLC; glioblastoma
MYCL1
Amplification
Transcription factor
SCLC, ovarian carcinoma
BCL2
Chromosome translocation
Antiapoptosis protein
B-cell lymphoma (follicular type)
CCND1
Amplification
Cyclin D, cell cycle control
Breast and other carcinomas
Chromosome translocation
B-cell lymphoma, parathyroid adenoma
BCR-ABL
Chromosome translocation
Chimeric nonreceptor tyrosine kinase
CML, ALL (T cell)
RET
Chromosome translocation
GDNF receptor tyrosine kinase
Thyroid cancer (papillary type)
Point mutation
Thyroid cancer (medullary type: germline mutations)
CDK4
Amplification
Point mutation
Cyclin-dependent kinase
Sarcoma, glioblastoma
MET
Point mutation
Hepatocyte growth factor (HGF) receptor
Renal carcinoma (papillary type: germline mutations)
SMO
Point mutations
Transmembrane signaling molecule in sonic hedgehog pathway
Basal cell skin cancer
CTNNB1 (β-CAT)
Point mutation, in-frame deletion
Transcriptional coactivator, links E-cadherin to cytoskeleton
Melanoma; colorectal, endometrial, ovarian, hepatocellular, and other carcinomas, hepatoblastoma, Wilms tumor
FGF4
Amplification
Growth factor (FGF-like)
Gastric carcinoma
PML-RARA
Chromosome translocation
Chimeric transcription factor
APL
TCF3-PBX1
Chromosome translocation
Chimeric transcription factor
Pre-B ALL
MDM2
Amplification
p53 binding protein
Sarcoma
GLI1
Amplification
Transcription factor
Sarcoma, glioma
TTG2
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access