Outline
Mechanisms of Human Gene Mutation
Genetic Alterations That Cause Cancer
Hereditary Cancer Risk Assessment: Genetic Counseling and Genetic Testing
Clinically Relevant Hereditary Syndromes
Hereditary Breast and Ovarian Cancer ( BRCA1 and BRCA2; Other DNA Repair Pathway Genes)
Lynch Syndrome (Hereditary Nonpolyposis Colon Cancer)
Less Common Inherited Cancer Syndromes Relevant to Gynecologic Oncology
Genetic Counseling and Genetic Testing for Hereditary Cancers
Referral of Gynecologic Cancer Patients for Hereditary Cancer Risk Assessment
Legal Aspects of Genetic Testing
Long-term Contact for Patients and Families With Hereditary Risk for Cancer
Key Points
- 1.
Cancer is not always inherited, but it is always genetic.
- 2.
The inherited aspect of cancer always needs to be considered and appropriate counseling and testing arranged.
- 3.
Genetic counseling and testing needs to be an integral part of all cancer management.
- 4.
Genetic counselors are necessary for management and treatment of inherited disease.
- 5.
Management is continuously changing, and communication among patients, their families, and the health care team is important.
Cancer is always genetic at the cellular level, but it is not always due to inherited risk. Overall, approximately 5% to 10% of cancers are clearly caused by inherited (germline) mutations. More recent data suggest that 10% to 25% of epithelial ovarian cancers are due to inherited risk. Mutations, if they occur in a gene or genes critical for cell growth, may allow a cell to reproduce in an uncontrolled fashion. This uncontrolled growth of a cell or tissue is known as a cancer. Cancer is seldom the result of a single mutation but is more commonly caused by a multitude of acquired mutations in many different genes, allowing uncontrolled cell division. The concept that cancer is the result solely of genetic or epigenetic changes is relatively new and comes from the past 4 decades of cancer research. Before the 1970s, the cause of cancer was essentially a black box, and its etiology was largely unknown. To some extent, it is still unclear, but we no longer believe that defective immunity, viruses, or metabolic errors directly cause cancer. These factors may contribute to the development of this disease, but the basis of cancer lies in an individual organism’s DNA and in that individual organism’s ability to express the genetic material in a normal fashion. This is simply related to the DNA sequences that make up that individual’s genome that has been inherited and altered through the course of normal living. Thus, cancer is a disease of abnormal DNA expression. Alterations or differences in the DNA sequences are known as mutations (changes in DNA such that a protein alteration occurs) or polymorphisms (changes in DNA that results in a subtle change in gene function and protein structure that do not cause disease). Polymorphisms are one of the factors responsible for normal population differences. There is a fine line between a mutation and polymorphism ( Figs. 19.1 and 19.2 ) (<1% but protein function remains).

An individual may inherit a genetic mutation or may be exposed to an environmental insult that predisposes to the development of a cancer. Such insults can occur from everyday living or as the result of repetitive exposure to an agent that can cause DNA damage such as known carcinogens or other environmental factors that cause DNA damage. In addition, any process that causes cells to divide, such as ovulation, can allow an opportunity for an error in DNA replication to be made. For instance, an individual prone to the development of ovarian cancer can reduce her risk of developing this disease by taking oral contraceptives. This will inhibit ovulation and decrease the damage to the surface of the ovary that could allow a mutation to occur. Recent hypotheses suggest that ovarian cancer may arise from the epithelium of the tubal epithelium. If this circumstance is true, then the etiology of oral contractive protection is not known.
Cancers, unlike many other inherited diseases, become apparent after there is an accumulation of several genetic injuries or mutations. In this respect, cancer is different from most inborn errors of metabolism because more than one genetic event is required to allow expression of the disease. One does not inherit cancers as one would inherit cystic fibrosis or sickle cell disease. One may inherit a mutation in a gene that predisposes one to the development of cancer, but most cancers are the result of the accumulation of somatic mutations that result from the process of normal living. Viruses, smoking, eating charcoal-broiled steak, or simply random mistakes made during DNA replication may cause these mutations. After all, a replicating cell must copy 3 billion base pairs—with each division, mistakes will occur.
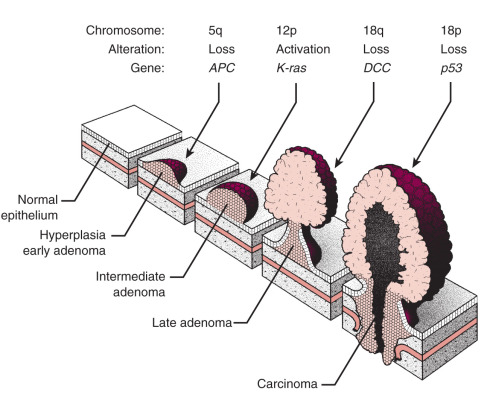
Progression from a normal cell to a malignant cell is the result of the accumulation of a series of mutations. This progression has probably best been demonstrated in colon cancer by Burt Vogelstein and colleagues ( Fig. 19.3 ). In this model, a series of mutations must occur that involve tumor suppressor genes and oncogenes; a benign tumor progresses to a malignant tumor as the necessary mutations occur during a prolonged and somewhat erratic interval.
Three broad classes of genes are involved in the development of cancer. These are tumor suppressor genes, oncogenes, and mismatch repair (MMR) genes. Tumor suppressor genes are responsible for making a product that inhibits cell growth. These types of genes are expressed in a recessive manner, and therefore both alleles need to be defective before the phenotype becomes apparent. Oncogenes are expressed dominantly and are usually responsible for a product that promotes cell growth. Overexpression of an oncogene protein results in uncontrolled cell growth. MMR genes are responsible for repairing DNA damage that results from loss of fidelity in normal DNA replication. If one of these genes does not function properly, errors in DNA can accumulate, and ultimately, some of these errors will occur in genes critical for cell control, resulting in uncontrolled cell growth. The development of cancers is therefore not the result of a single error or insult but rather the accumulation of genetic errors over time.
Mutations are a means of evolution. The paradox of life is that mutations can be responsible for an individual organism’s death in the form of cancer or metabolic error, but other mutations can account for the evolution of the species. Therefore, mutations can be good, bad, or even neutral.
Genetic Alterations in Cancer
Genetics is not a new field and has been a study for several centuries from Mendel in 1865 to the present. Johannsen first coined the term gene in 1911 as it applied to the unit of a hereditary characteristic. This was further refined to the one gene–one enzyme concept in the 1940s and put forth by Tatum and Beadle. These concepts have been clearly defined in Drosophila but apply equally to humans and other organisms; after all, all living things are defined by their DNA. Humans have more DNA than other organisms and are therefore more complex, but the DNA behaves the same way in all organisms.
The one gene–one enzyme concept that developed from the ideas expressed by Tatum and Beadle can be summarized as follows:
- 1.
All biologic processes are under genetic control.
- 2.
These biochemical processes are expressed as a series of stepwise reactions.
- 3.
Each biochemical reaction is ultimately under the control of different single genes.
- 4.
Mutation of a single gene results in an alteration of the cell to carry out a single chemical reaction.
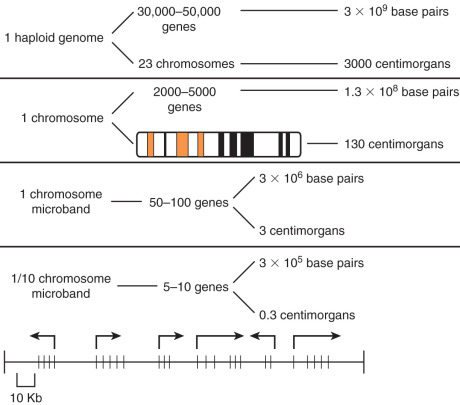
The one gene–one enzyme concept has since been refined and extended to cover proteins that are not enzymes. Now further information suggests that some genes combine with others to form unique proteins, indicating that a few genes may interact to form more than one protein per gene. The human genome is estimated to contain about 3 billion base pairs and was thought to have between 50,000 and 100,000 genes. Now that the human genome has been sequenced, we have discovered that there are only about 30,000 genes. These genes are stored on linear strands of DNA and interact much more than we previously believed. The genes are not continuous sequences of DNA but consist of coding sequences called exons interrupted by noncoding sequences called introns. Most of the DNA is composed of intronic DNA that is usually not expressed in any form. An approximation of the size of the human genome and its organization can be seen in Fig. 19.4 . The functional length of the human chromosome is expressed in centimorgans (cM). A cM is the distance over which there is a 1% chance of crossover during meiosis and is really a measure of “genetic distance.” Linkage studies indicate that the human genome is about 3000 cM. The average chromosome contains about 1500 genes in 130 million base pairs. Fig. 19.4 shows the estimated physical and functional size of the genome. Whereas the physical size is estimated in base pairs, the functional size is estimated in centimorgans. Much, if not most, of the human genome is composed of noncoded DNA and is therefore not expressed.
DNA is transcribed into RNA that is translated into protein. Therefore, the sequence of DNA base pairs ultimately determines the sequence of the functional proteins within the cell. All cellular functions are expressed in this manner ( Fig. 19.5 ):
DNA→RNA→Protein
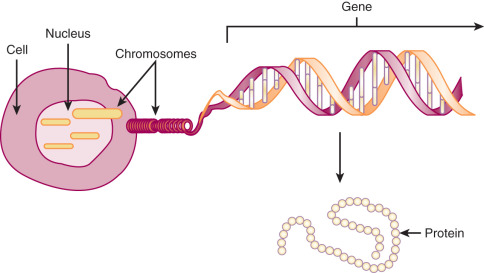
Humans and other mammals may be more complex than some other organisms in that pieces of genes may combine with parts of other genes to make a totally new protein. Hence, we may essentially increase the number of genes expressed without really increasing the amount of functional DNA or the number of actual genes. Overall, less than 30% of our genetic material is expressed.
Mechanisms of Human Gene Mutation
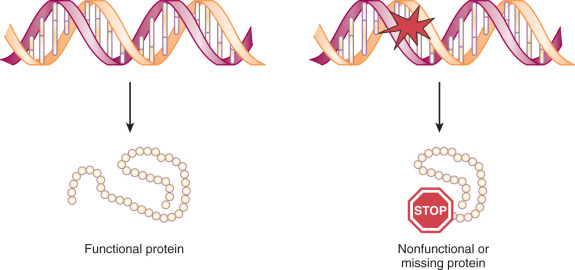
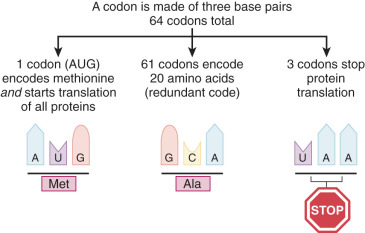
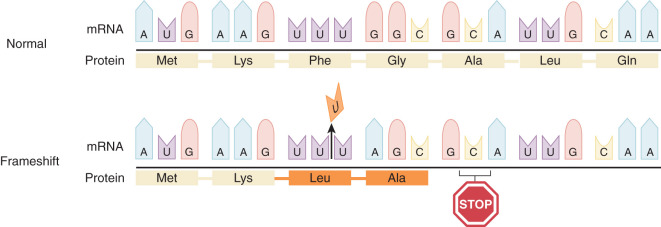
All cancers result from alterations in DNA structure, leading to changes in the proteins responsible for cellular function. All living organisms use the same 20 amino acids as building blocks for proteins. The translation of a nucleotide sequence from DNA to protein is dependent on a triplet code of nucleotides. Each of these triplet codes is called a codon. Some codons have very special functions. The code AUG codes for methionine, and this starts the initiation of synthesis (translation) of every protein. The code UAA is a stop codon and ends translation of a protein ( Fig. 19.6 ). Much of the data available about genetic alterations in cancers come from studies other than of gynecologic malignant neoplasms. These studies often involve hematologic malignant neoplasms such as lymphomas and leukemia because pure subsets of cells are easily obtained. More recently, we have been able to improve our study of solid carcinomas because of our ability to study pure tumor preparations, single out cells, and isolate the DNA with microdissection and laser capture techniques. Using this technique and techniques like it, we can dissect out a single cancer cell, replicate its DNA, and evaluate the mutations that occur in this single cell or group of cells. Fig. 19.7 demonstrates the heterogeneity of cancer and why there is a need for these special techniques. Cancer is the result of clonal expansion, but as the various cells replicate in an uncontrolled manner, they continue to mutate. Initially, cancers usually have a single clonal origin. As the cancer progresses, mutations occur, and therefore some cells may become genetically different from the original cancer. Hence, the term mutator phenotype is often applied to cancers.
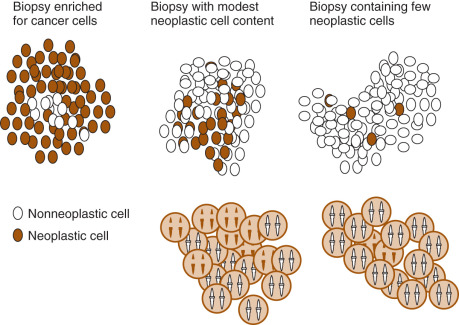
A wide number of and various types of mutations have been described. These mutations are fundamental to the understanding of cancer and of human evolution. Although many types of mutations can occur, some common ones are listed in Table 19.1 . Multiple searchable databases are publicly available that provide a comprehensive listing of gene specific mutations reported in the somatic cancer tissue as well as germline (inherited) mutations. Examples of these databases include The Human Gene Mutation Database (HGMD) ( http://www.hgmd.cf.ac.uk/ac/index.php ), Clinvar ( http://www-ncbi-nlm-nih-gov.easyaccess2.lib.cuhk.edu.hk/clincar ), and COSMIC (Catalogue of Somatic Mutations in Cancer; http://www.cancer.sanger.ac.uk/cosmic ). These databases are periodically updated as most researchers and commercial laboratories publicly share their data. Illustrations of some common mutations can be seen in Figs. 19.8 to 19.13 .
|

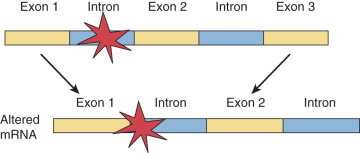
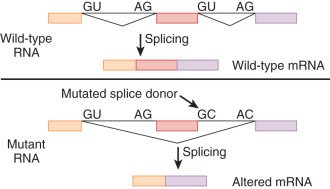
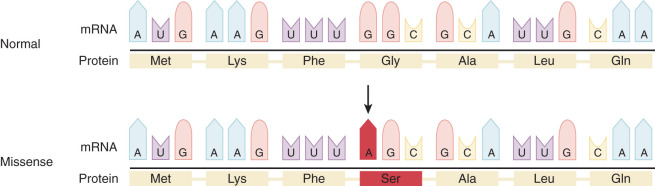
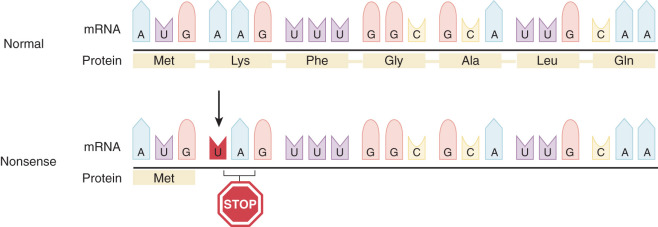
Single Base Pair Substitutions and Point Mutations
DNA replication is the result of an accurate yet accident-prone process. The final accuracy of DNA replication depends on the fidelity of the initial process and the ability of subsequent repair processes to correct any mistakes. Point mutations lead to single changes in the DNA sequence. These single mutations can have very different effects on the reading of a protein, as demonstrated in Figs. 19.10 and 19.11 . If this mutation is in a regulatory portion of a gene, loss or alteration of regulation of gene expression can occur. If the mutation is in a coding portion of the gene, an altered protein can be formed. HGMD is one of several catalogs of reported point mutations that can be obtained from the Internet.
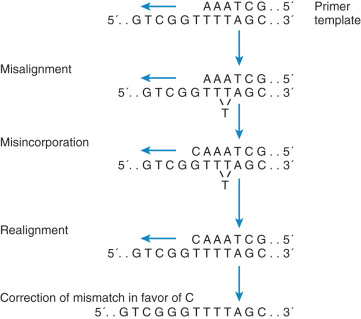
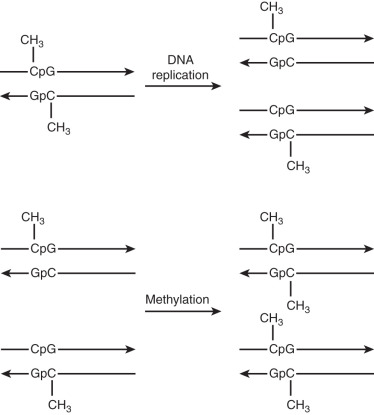
One of the most common mutations as the result of substitution occurs in CpG dinucleotides (3′ cytosine-guanine 5′). Data show that these transversions account for about 30% of point mutations. The fidelity of this process is related to the efficiency and accuracy of the DNA polymerase responsible for replication. Other types of these small nucleotide substitutions that can also occur are messenger ribonucleic acid (mRNA) splice junction mutations, translational mutations, and single deletions. Alternatively, mismatches can occur by slipped mispairing, which leads to single base pair mutagenesis and deletion.
Deletion or insertion of a single nucleotide may be secondary to DNA mispairing during replication. This type of mutation often occurs in runs of identical base pairs and is the result of slippage of a single base pair ( Fig. 19.14 ). This mechanism is similar to larger deletions caused by this same mechanism. The significance of this type of mutation to human malignant neoplasia is as yet undefined. As more information becomes available about mismatch repair, we should have a better estimate of the significance of this type of mutation to cancer formation. However, this slippage is common and similar to the errors found in individuals who have an inherited cancer syndrome, hereditary nonpolyposis colon cancer (HNPCC), or Lynch syndrome.
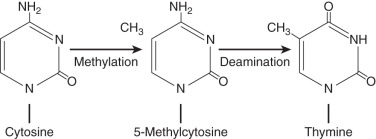
CpG dinucleotides are very susceptible to mutations. Methylation of cytosine results in a high level of mutation because of its propensity to undergo deamination. This then forms thymidine and therefore can result in a change of gene expression. The evolution of the human genome seems to indicate a progressive loss of CpG islands because of this transition. The frequency of these islands is about 20% to 30% of the predicted frequency, probably because of the progressive loss of CpG dinucleotides. This excess of transition from cytosine to thymine (C to T) transitions was first reported by Vogel and Rohrborn in a study of the mutations responsible for variants of hemoglobin. Others now also have shown that CpG islands are associated with a high rate of transition, supporting the thought that these mutations play a significant role in human disease ( Fig. 19.15 ).
Larger Deletions
Gene deletions are responsible for more than 150 inherited diseases. Deletions can range from a few base pairs to several hundred kilobases and are often classified by size. These are also nonrandom in that certain sequences appear to be more prone to deletion than others are. This nonrandomness is apparent when one evaluates genetic disease. Spontaneous deletions often occur in the same sequences of various genes, indicating that some sequences of DNA are more prone to deletion than others.
Deletions occur when there is homologous but unequal recombination between gene sequences. Similar sequences in the human genome can cross over during mitosis or meiosis, resulting in a shortened portion of the gene sequence. Long areas of homology (homology boxes) are thought to be the most likely to have this type of mutation. Repetitive DNA sequences are particularly susceptible to deletions because this can allow slippage. This mechanism is called homolog or unequal recombination between repetitive sequence elements. The most common repetitive sequence is the Alu repeat (up to 106 Alu sequences are in the human genome). Alu repeats are characterized by an average spacing of 4 kilobases and 300–base pair length separated by a short A-rich region. The Alu element is 70% to 90% homologous to the consensus sequence. The 3′ homology (homology boxes) is thought to be the most likely to have this type of mutation. Repetitive DNA sequences are particularly prone to mutation.
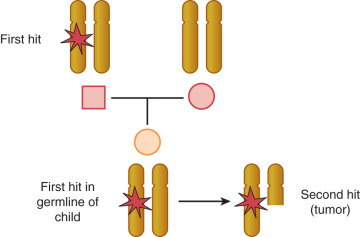
There are more than 400 gross gene deletions recorded in the HGMD and 3000 or so smaller gene deletions. Almost 2500 gene deletions of 20 base pairs or fewer have been identified. These can be the result of deletion of repeats or a part of the excision repair process. Deletion is not random; particular sequences are more prone to deletion and often contain regions involving guanine or other repeats. The application of the techniques of molecular biology, such as gene cloning, in situ hybridization, restriction endonuclease mapping of genetic sequences, and polymerase chain reaction analysis of gene transcription, has led to the conclusion that a given chromosome abnormality may be associated with a variety of neoplasms and that a given oncogene may be activated in a variety of human cancers. The most common defects in solid tumors are deletions in specific gene sequences, observed as a loss of part of a banding region or the loss of heterozygosity of a specific genetic allele. Deletion of genetic material in a cancer cell suggests loss of function that regulates cell proliferation and differentiation. Many human solid tumors have been shown to have some type of chromosome deletion. The Tp53 tumor suppressor gene–containing region of chromosome 17p is deleted or mutated in a wide variety of human cancers. The fact that there is such commonality among cancer cell types in the loss of chromosome material suggests that these regions contain genes coding for regulating functions of a wide variety of cell types. Induction of the malignant neoplastic process must involve at least two hits when a tumor suppressor gene is involved. This implies that two mutations or deletions in each of the two alleles are necessary before the effect can be expressed. This is the two-hit theory proposed by Knudson. The possibilities for the two hits necessary to express a recessive gene can be seen in Fig. 19.16 . This explained why some people could inherit a predisposition for a malignant disease and also raised the question of whether sporadic cancers could have a genetic basis. In a genetically predisposed cell, the remaining single normal allele may be sufficient to maintain normal growth and regulation; a second deletion or mutation is required to inactivate the remaining normal allele.
Insertions
Most insertions appear to be small, between one and a few base pairs. This phenomenon has been studied extensively, and these insertions often appear to be nonrandom in character. Predisposing factors lead to these insertions. These insertions can be due to slipped mispairings, or they may be mediated by inverted repeat sequences or by symmetric elements. These sorts of DNA sequences are particularly prone to mutation.
Less common are large insertions. The largest insertion known is the 220-kilobase segment inserted into the DMD gene. Alu and LINE (long interspersed nucleotide elements) sequences are especially susceptible to insertion and deletion. Mutations are nonrandom, and some DNA segments are more susceptible to mutation than others are. For example, the myc gene is especially susceptible to insertions; a 50–base pair LINE element has been noted to be inserted in this gene in breast cancer cell lines. The fact that insertional mutations may not be random is underscored by the finding of Fearon and Vogelstein, who reported 10 independent examples of insertions within the same 170–base pair intronic region of the DCC gene involved in the progression of colorectal cancers.
Duplications
Duplication of small parts of whole exons, or even of whole chromosomes, has played a large part in the development of cancer. This phenomenon also has a large role in the evolution of the human genome. Again, the paradox is that mutations are sometimes good and sometimes bad. Duplication of larger segments of DNA may lead to gene amplification. Gene amplifications can occur in many ways. A gene that is usually present in a single copy in the normal cell may be duplicated or may undergo a small increase in the copy number. Another type of amplification may involve a 10- to 100-fold increase in copies of a genetic locus containing key regulatory genes. A third type of amplification can result in the duplication of whole chromosomes. One can envision how amplification has been important over the course of evolution. We have 23 pairs of chromosomes, and all other organisms have fewer chromosomes. This is due to millions of years of evolution resulting from innumerable insertions and duplications.
Inversions
Inversions are perhaps the least common type of mutation. In this mutation, a long segment of DNA forms a loop and alters its direction; the 3′ end now becomes the 5′ end. These segments can obviously be large or quite small. The most important inversion event in human disease involves the factor VIII gene, causing severe hemophilia A. An MSH2 inversion has been reported in exons 1 to 7 in Lynch syndrome and is one of the few cancer-associated inversions.
Translocations
More than 100 common translocations have been observed in malignant cells. Many of these occur consistently in certain specific cancer types, which argues that these areas are involved in the malignant process of the cell. Some of the translocations may be secondary events in the evolution of more aggressive phenotypic changes. The inherent genetic instability of malignant cells leads to further karyotypic abnormalities as the disease progresses, reflecting additional genetic alterations that increase growth potential. Evidence that malignant transformation does not usually result from a single translocation event comes from the study of patients with ataxia-telangiectasia, who are at high risk for leukemia. These patients have lymphocytes with a characteristic translocation present for many years before a malignant change develops.
A common finding in cancers is some form of genetic instability. This is most clearly manifested in Lynch syndrome (or HNPCC). Lynch syndrome is characterized by genetic instability at microsatellite sequences and has been linked to several MMR genes. Mutation rates in cells that are positive for MMR defects are orders of magnitude higher than MMR intact cells.
Cancers in patients with hereditary cancer syndromes may serve as a model for sporadic disease because mutations that occur in the sporadic tumors are often very similar. The same genes and sequences seem to be targeted. For instance, Krawczak and Cooper demonstrated that the bulk of single base substitutions in the Tp53 gene in sporadic cancers strongly resemble those found in inherited cancers.
Cancer Epigenetics
Epigenetics refers to modification of gene expression not specifically related to changes in the DNA sequence of a gene. This is a concept of differential expression of the genotype based on factors outside of the genome. It is interesting that these differences can be inherited and appear to be quite stable. Epigenetic changes can involve chromatin structure; histone modification; changes in transcriptional activity; and, most easily measured in mammals, changes in methylation of CpG loci.
Methylation is the only known covalent modification of DNA in normal mammalian cells. This change occurs at the fifth position of cytosine at 5CG-3′ dinucleotides. About 80% of these types of dinucleotides are methylated and therefore are probably important in cell control and gene expression. Generalized hypomethylation is present in colorectal cancers and endometrial cancers early on in the development of these diseases, but the exact cause and effect of this observation is unclear. Presumably, hypomethylation allows some genes to be overexpressed. Alternatively, some regions on some genes, particularly the promoter region of MMR genes, can cause gene silencing ( Fig. 19.17 ). This allows a tumor suppressor gene to be silenced, thus promoting the development of that cancer. Changes in methylation status are not clonal. Hence, some cancers can have different gene expression in the same cancer because of differential methylation. Changes in methylation status could also cause aberrant chromosome condensation during division and result in abnormal chromosome segregation. Therefore, epigenetics can affect tumor behavior in several ways.
Genomic Imprinting and Cancer
Genomic imprinting is a modification of DNA that leads to a different expression of the gene depending on the parent of origin. This may be passed from generation to generation. However, the specific expression of these modifications may increase, decrease, or remain stable from generation to generation ( Fig. 19.18 ). These epigenetic alterations appear to occur commonly in human malignancies and because of their unique properties may lead to novel forms of therapy.
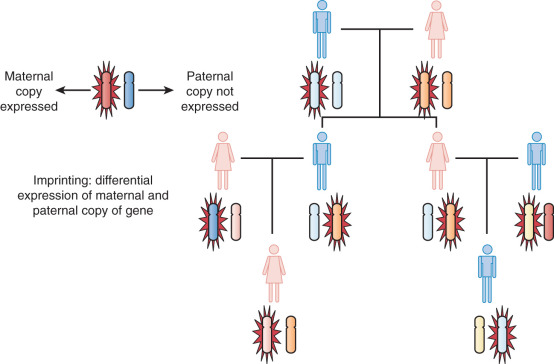
Assumptions are made in Mendelian genetics, which we apply to human disease, that the maternal and paternal alleles are equivalent. We also assume that both copies of the gene are necessary for normal function. This is not true with respect to genomic imprinting. An example of imprinting is the differences noted between the mule and the hinny. The mule is the cross between a maternal horse and a paternal donkey. It is much larger than the hinny, which is a cross between a maternal mule and a paternal horse. Although these two have the same genomic equivalent, they are distinctly different, indicating differential expression of maternal and paternal genes.
Examples of genomic imprinting and cancer are the hydatidiform mole and the teratoma. The hydatidiform mole is composed of paternal chromosomes, and the teratoma is composed of only maternal chromosomes. These examples demonstrate that not only does it take 46 chromosomes to make a human being, but there must also be a balance between maternal and paternal elements.
Genetic Alterations That Cause Cancer
Oncogenes
The past 25 years have contributed significantly to our understanding of the molecular mechanism of cancer and have identified three types of molecular aberrations that can lead to cancer: dominant transforming genes called oncogenes, recessive transforming genes called tumor suppressor genes, and genes responsible for repairing DNA errors (MMR genes). Many oncogenes have been isolated as forms of proto-oncogenes acquired by RNA tumor viruses. We have known for many years that viruses can cause malignant tumors in animals. The link noted in animals spurred a great deal of research aimed at identifying the cancer-causing genes carried by the viruses and finding the human genes that were affected. These investigations surprisingly revealed that the genes implicated in malignant disease were often altered forms of viral genes. These were probably acquired when the virus infected the animal and then moved on to other animals. At other times, the viruses activated host genes that were normally quiescent. The normal versions of these pirated and activated genes, now termed proto-oncogenes , carried codes specifying the composition of proteins that encourage and stimulate cell replication. These growth-promoting genes come in various forms. Some specify the amino acid sequences of receptors that are found on the cell surface and bind to molecules known as growth factors. When bound by such factors, the receptor issues an intracellular signal that ultimately causes cells to replicate. Other growth-promoting genes code for proteins that lie inside the cell and govern the propagation of intracellular growth signals. A third group encodes proteins that control cell division and are under nuclear control. These oncogenes can be activated via several mechanisms. The oncogene can be amplified, and many copies of the gene can become activated. Occasionally, the gene may be translocated to another chromosome, where, under the influence of another promoter, it promotes uncontrolled growth.
The three classes of oncogenes are controlled differently. The first comprises a group of peptide growth factors and their receptors. Included in this group are the peptide growth factors and their receptors such as epidermal growth factor receptor (EDGFR) and platelet-derived growth factor (PDGFR). These peptide growth factor receptors likely serve as stimulatory cofactors rather than the actual force that drives the malignant process. Targeted therapy is becoming a reality as our understanding of these cancer-promoting entities increases. Unique molecules are aimed at gene products or proteins activated by these factors. Another class of oncogenes comes from non–membrane-traversing extranuclear growth factors. These include G-proteins and the ras gene family. Finally, there are oncogenes of the nuclear regulatory variety such as myc. A schematic of oncogenes representing their function can be seen in Fig. 19.19 . A more detailed description of targeted therapy can be found in Chapter 18 .
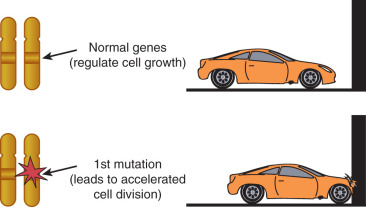
The discovery that viral genes had human counterparts introduced the intriguing possibility that human cancers, including the majority not caused by viruses, might originate from mutations that convert useful proto-oncogenes into harmful genes—that is, oncogenes. Consistent with this notion, studies indicated that alteration of just one copy, or allele, of these proto-oncogenes was enough to transform and render cancerous some types of cells growing in culture. Such dominant mutations cause cells to overproduce a normal protein or make an aberrant form that is overactive. In either case, the result is that stimulatory signals increase within the cell even when no such signals come from the outside.
It is ironic that research on animal RNA tumor viruses (retroviruses) having no ability to cause human cancer provided the first key to uncovering the identity of some oncogenes and the verification that they actually exist. These retroviruses, which infected chickens, rodents, cats, and monkeys, were extremely tumorigenic in that infected animals often showed tumors on initial exposure to the virus. One of these viruses, the Rous sarcoma virus of a chicken, was found to carry a specific gene that it used to transform infected cells to a malignant state. This type of transforming oncogene was termed a viral oncogene . A single oncogene carried onto a chicken cell by a Rous sarcoma virus was able to derail and redirect the entire metabolism of the cell, forcing it to grow in a malignant fashion.
In 1976, Varmus and Bishop found that the oncogene in the Rous sarcoma virus was not a viral gene at all; instead, it arose directly from a preexisting cellular gene that had been captured by an ancestor of the Rous sarcoma virus. After it was captured, this gene was used by the Rous sarcoma virus to transform mammalian cells. The early ancestor of the Rous sarcoma virus was capable of replicating in infected cells but was unable to transform them; it instantly gained tumorigenic potency by kidnapping this normal cellular gene, the proto-oncogene. In the end, this work by Varmus and Bishop revealed much more about the cell than it did about the Rous sarcoma virus because it pointed to the existence of a gene residing in a normal mammalian genome that possessed potent transforming ability when appropriately activated, in this case by a retrovirus. Here was clear proof of the presence of at least one gene in the normal cellular DNA that might serve as one of the target genes activated by nonviral carcinogens of the retrovirus; such nonviral agents might also activate this proto-oncogene, converting it to a powerful oncogene. A cell carrying such a mutant gene might, in turn, respond to this damage by launching a program of deregulated growth and thus become a cancer cell.
The information uncovered concerning retroviruses and oncogenes provided little immediate comfort to investigators interested in the origins of human cancer. Retroviruses, similar to Rous sarcoma virus, never infected humans and therefore could not act to mobilize human proto-oncogenes. The possible connection came from a notion that many such proto-oncogenes could be activated through an alternative route. Changes in the DNA sequence created by chemical or physical rearrangement might substitute for the virus. Mutations induced by these chemical or physical conditions in the genomes of target cells in one or another tissue might be as effective as retroviruses in activating latent carcinogenic potential in the proto-oncogenes. By the early 1980s, these suspicions were validated: mutated genes (proto-oncogenes) were found in human tumor genomes. In each case, a change in the sequence of the gene was identified as being responsible for converting a proto-oncogene into an active oncogene. For example, a ras oncogene in one human bladder carcinoma was found to have arisen through a single base change that altered the DNA sequence of a precursor proto-oncogene; myc oncogenes arose through gene amplification in various malignant neoplasms.
Next, researchers tried to ascertain how these genes succeeded in transforming cells. A simple theme emerged that makes it possible to understand and explain the mechanism of action of most, if not all, of the oncogenes. This comes through the understanding of how cells regulate their own growth. The growth and division of a normal cell residing in a particular tissue is controlled largely by its surroundings. A normal cell rarely, if ever, decides its own rate of proliferation; rather, it responds to the signals or messages from surrounding cells. These messages, which may carry growth-stimulatory or growth-inhibitory information, are conveyed by growth factors released by surrounding cells, traverse the intercellular space, and bind to the cell surfaces. These cells then respond to these growth signals by activating their synthetic machinery, copying their DNA, and dividing. A normal cell will never commit itself to such a growth program without having been simulated by these external signals. Each cell possesses complex machinery that enables it to receive these signals, process them, and launch a growth program. The machinery consists of an array of proteins that function to acquire growth-activating signals and transmit them throughout the cell. These proteins include the following:
- 1.
Cell surface receptors that recognize the presence of growth factors in the extracellular space and transmit signals into the cell’s interior
- 2.
Cytoplasmic signal transducers that become activated by these receptors and then pass signals farther into the cell
- 3.
Nuclear transcription factors that are activated by the cytoplasmic signal transducers and in turn respond by activating entire banks of cellular genes
These activated gene banks together orchestrate the cell’s growth program; they detail events that, acting in concert, enable the cell to grow and divide. Proto-oncogenes encode many of the proteins in this complex signaling circuitry that enable a normal cell to respond to exogenous growth factors. Oncogenes participate in this signaling circuitry by selecting aberrantly functioning versions of the components of this circuitry. Oncogene proteins succeed in activating these signal circuits even in the absence of stimulation by extracellular growth factors. In doing so, they force a cell to grow even when its surroundings do not contain some of the clues that are normally required to provoke growth.
Other researchers suggest that mutations in at least two proto-oncogenes have to be present and that only certain combinations of mutations lead to malignant change. These findings suggest that individual oncogenes, although powerful controllers of cell metabolism, are not capable of causing malignant neoplasms by themselves. Thus, oncogenes cannot explain most cancers by themselves. This view was strengthened by the discovery of more than a dozen different oncogenes in human tumors ( Table 19.2 ). However, on careful evaluation, only about 20% of tumors turned out to carry expected alterations. None of the tumors had pairs of cooperative alterations sometimes found in cultured cells. It also appeared that the inherited mutations responsible for predisposing people to cancer were not oncogenes. The concept of a recessive-type antioncogene was conceived. This type of gene is called a tumor suppressor gene. This class of gene appears to be equally important in the development of cancer.
| Oncogene | Associated Tumors |
|---|---|
| hst | Gastric cancer |
| Neu-erB-B2 | Breast, ovary, and gastric cancers |
| trk | Papillary thyroid and colon cancers |
| Ha-ras | Bladder cancer |
| Ki-ras | Lung and colon cancers |
| N-ras | Leukemias |
| gsp | Pituitary tumors |
| raf | Gastric cancer |
| met | Osteosarcoma |
| abl | Leukemia and lymphoma |
| myc | Lymphomas and carcinomas |
| N-myc | Neuroblastoma |
| L-myc | Small cell lung cancer |
| bcl-2 | Follicular and undifferentiated lymphoma |
| mas | Breast cancer |
| ret | Papillary thyroid cancer |
Tumor Suppressor Genes
The direct identification of tumor suppressor genes has been more difficult than the identification of oncogenes. Tumor suppressor genes were first conceived of in a theoretical sense long before any were actually identified. Harris was the first to demonstrate that the malignant characteristics of a cell could be suppressed when malignant cells were fused with nonmalignant cells. Furthermore, loss of portions of chromosomes can be associated with malignant transformation, and transfer of a normal chromosome can reverse these traits into the malignant cells. These data suggested a recessive type of control in normal cells. A schematic of a tumor suppressor gene can be seen in Fig. 19.20 .
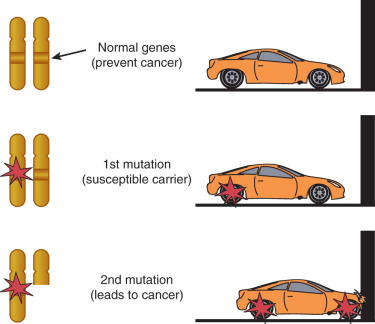
The first tumor suppressor gene cloned was the RB gene; it is the defective gene in retinoblastoma. Knudson extensively studied the epidemiology of retinoblastoma. Most cases of retinoblastoma are sporadic, but some cases appear to be clustered around families. He noted that cases of familial retinoblastoma were more likely to be bilateral or multifocal. On the basis of these data, he developed the two-hit hypothesis that bears his name (see Fig. 19.16 ). That is to say that two mutagenic events are required for retinoblastoma to develop. In patients with inherited disease, the first hit was inherited in one allele of the RB gene in the germline. Although one abnormality was not enough for development of the disease, there was an increased likelihood that the second allele would be damaged during eye development. The hypothesis of two mutational events developed by Knudson applies specifically to tumor suppressor genes. This is the basis of most inherited gene models.
Tumor suppressor genes have proved difficult to identify, and many techniques have been used to locate such genes within the human genome. Cytogenetic studies provide some clues to the location of tumor suppressor genes. A significant percentage of patients with retinoblastoma or some patients with Wilms tumor have large deletions in 13q14 or 11p13, respectively. These are the regions of the respective tumor suppressor genes responsible for each malignant neoplasm. Significant loss on 5q is associated with adenomatous polyps of the colon. This can be measured on a gel during electrophoresis and is called loss of heterozygosity. Although recurrent loss of a chromosome region associated with a specific malignant neoplasm is highly suggestive that this region is important in the development of the cancer, it is not sufficient to prove that there is a tumor suppressor gene in this region. Fig. 19.16 shows the possible combinations of loss that would lead to the expression of a tumor suppressor gene. Use of genome-wide linkage scans can further localize the presence of such a gene. Much smaller areas of deletion can be detected by molecular techniques such as pulsed-field electrophoresis. Now we can also easily and cheaply sequence long segments of the genome as these technologies improve.
When scientists were first trying to identify the regions of loss, they used DNA probes for specific regions of chromosomes. If allelic loss is noted, this is called loss of heterozygosity; if this loss is disproportionate relative to the general population, this suggests a tumor suppressor gene in the region specific for the probe. An example of loss of heterozygosity can be seen in Fig. 19.16 . Here, a region on 10q in endometrial cancers indicates the existence of a possible tumor suppressor gene. By use of various markers, a precise location of the tumor suppressor gene can be identified. A contig of the region must then be built and the region sequenced to look for specific genes. Many regions of loss have been identified by this technique. There are many more candidate regions than actual tumor suppressor genes.
The tumor suppressor gene Tp53 is a gene associated with many cancers that was identified by searching for loss of heterozygosity. Several other tumor suppressor genes have been identified since. This is almost certainly only the tip of the iceberg, and many more tumor suppressor genes are likely to be discovered as investigators learn more about cancer cell genetics and as the potential of the Human Genome Project is realized and new genetic testing technologies continue to bring genetic testing to the forefront of medicine. Several generalizations can be made, and a comparison with the activation of oncogenes is constructive. Mutations in oncogenes are gain of function events and lead to increased cell proliferation and decreased cell differentiation. Oncogene mutations do not appear to be inherited through the germline. In contrast, tumor suppressor gene inactivations are loss of function events, usually requiring a mutational event in one allele followed by a loss or inactivation of the other allele. This loss of gene function leads to loss of cellular control and unchecked growth. Tumor suppressor genes are recessive, and mutations may be inherited as a germline mutation. Somatic mutations may occur in both types of genes and accumulate throughout life. Comparison of oncogenes and tumor suppressor genes is made in Table 19.3 . Common tumor suppressor genes are listed in Table 19.4 .
| Characteristic | Oncogene | Tumor Suppressor Gene |
|---|---|---|
| Number of mutational events in cancer | One | Two |
| Role of mutation | Gain of function (“dominant”) | Loss of function (“recessive”) |
| Germline inheritance | No | Yes |
| Somatic mutations | Yes | Yes |
| Genetic alterations | Point mutations, amplifications, gene rearrangements | Point mutations, deletions |
| Effect on growth control | Activates cell proliferation | Negatively regulates growth-promoting genes |
| Result of gene transfection | Transforms cells to a partly malignant behavior | Suppresses malignant phenotype |
| Gene | Gene Product | Tumor Associations |
|---|---|---|
| RB1 (13q) * | 110-kDa nuclear hypophosphorylated protein, negative cell cycle regulator | Retinoblastoma |
| Osteosarcoma | ||
| Small cell lung cancer | ||
| Soft tissue sarcoma | ||
| Breast cancer | ||
| Bladder cancer | ||
| p53 (17p) | 53-kDa sequence-specific DNA-binding protein and transcriptional activator | Li-Fraumeni syndrome Most common alteration in human cancer |
| DCC (18q) | 1447–amino acid transmembrane protein with homology to known adhesion molecules; role in terminal cell differentiation | Colorectal cancer |
| APC (5q21) | 2843–amino acid protein that interacts with membrane-associated cadherin-catenin complexes and with microtubules | Familial adenomatous polyposis |
| Gardner syndrome | ||
| MTSI (9q21) | 148–amino acid protein inhibitor of cyclin-dependent kinase-4 | Familial melanoma Bladder cancer |
| BRCA1 (17q) | 1863–amino acid protein with zinc finger–like domains suggesting function as a transcriptional factor | Breast cancer |
| Ovarian cancer | ||
| BRCA2 (13q) | Possibly BRUSH I | Familial breast cancer |
| VHL (3p) | Protein with short homology to a glycan-anchored membrane protein of Trypanosoma brucei ; no function assigned | Pheochromocytoma |
| Renal cell cancer | ||
| Pancreatic cancer | ||
| Hemangioblastomas of CNS and retina | ||
| WT1 (11q) | 50-kDa gene, related to the early growth response gene; encodes four 46- to 49-kDa proteins, which appear to function as DNA-binding transcriptional repressors | Wilms tumor |
| NF1 (17q11.2) | Neurofibromin (~2500 amino acids); probably negative regulator for p21 ras | von Recklinghausen neurofibromatosis |
| NF2 (22q12) | Schwannomin (~600 amino acids), regulator of cellular response to external environment | Neurofibromatosis type 2 |
Apoptosis
Apoptosis means programmed cell death and refers to the “intentional” induction of cell death. Apoptosis is important in the growth and development of an organism because as an organism matures and differentiates, cells must die to give way to more differentiated and specialized cells. If a cell does not die and becomes immortal, a tumor or cancer can result. Apoptosis was first described in the 1970s, but only recently have scientists begun to realize the importance of this phenomenon in organism development, differentiation, and cancer formation. Excitement about this process has been driven by the finding that apoptosis is controlled at the molecular level by genes associated with malignant change (ie, oncogenes, proto-oncogenes, and tumor suppressor genes). It is also clear that many of these same controlling elements and pathways are active during development. Many believe that understanding the control of the apoptotic process is essential to understanding the control of the developing organism and control of senescence. Loss of these aspects of cellular control could lead to cancer.
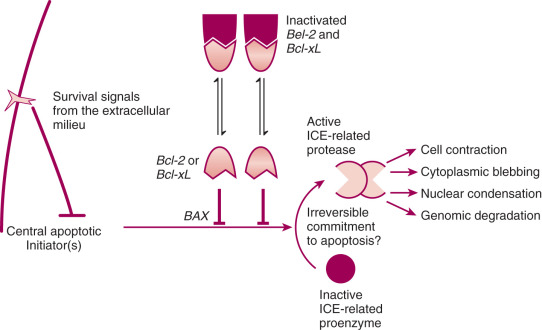
Apoptosis is a distinct mode of cell death that is responsible for the deletion of cells in normal tissues; of note, it also occurs in pathologic conditions. The process of apoptosis is characterized histologically by cell contraction, blebbing of the cell membrane, and condensation of the nucleus. Apoptotic bodies are formed that contain intact organelles; surrounding cells eventually phagocytose these. There is no associated inflammatory response. Cell swelling and disintegration of the cell components characterize necrotic cell death, and there is a marked inflammatory response. On a molecular level, apoptotic death causes cellular enzymes to autodigest the genome into little fragments, and this can be identified on polyacrylamide gels by multiple regular fragments seen as a DNA ladder.
Apoptosis is important in the development of the normal organism, and it is important in the development and growth of cancers. It occurs spontaneously in malignant tumors, often markedly retarding their growth, and it is increased in tumors responding to radiation, chemotherapy, heat, and hormone damage. In cancer, apoptosis appears to be a mechanism for deleting cells from the population that have sustained carcinogenic DNA damage; however, when apoptosis of such cells is blocked or inhibited by mutations in genes that help control this process, such as BCL2 or Tp53, these cells are suddenly free to continue replicating and propagating their mutations. This genetic instability may be an early step in the development of cancers. Many of the current cancer treatments, such as radiotherapy and chemotherapy, kill cells by the production of DNA damage. Mutations in BCL2 and Tp53 may then influence the effectiveness of these therapies through their ability to inhibit cell death.
The primary importance of apoptosis related to cancer and cancer treatment lies in its being a regulated phenomenon subject to stimulation and inhibition. Although little is known about how to establish therapeutic agents to affect its initiation, it seems reasonable to suggest that greater understanding of the process of apoptosis might lead to the development of improved treatment possibilities. Inhibitory mechanisms such as BCL2 proto-oncogene expression may be implicated in the development of resistance to therapeutic agents and may contribute to tumor growth and perhaps to oncogenesis by allowing the inappropriate survival of cells with DNA abnormalities. It is likely that other inhibitory mechanisms will be identified, and a better understanding of the apoptotic process may lead to novel treatment regimens by allowing us to control cell death. Apoptosis is not simply a description of cell death, nor is it a spurious trend in the biology literature. It is a fundamental process and is controlled at the molecular level; as such, it can be understood and manipulated. Fig. 19.21 demonstrates how apoptosis may occur.
Mismatch Repair Defects
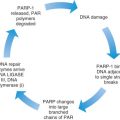
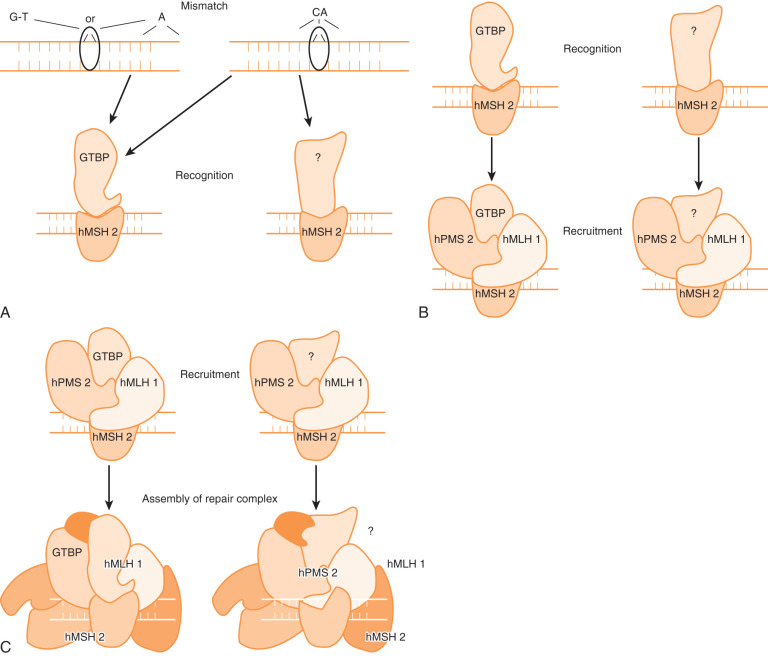
A number of mechanisms are involved in the correction of DNA changes that result from exogenous or endogenous mutagenic agents. These mechanisms include base excision repair, nucleotide excision repair, and DNA mismatch repair. The MMR system repairs mistakes that are made in the course of normal cellular division. These mutations often occur in regions of multiple repeats because these regions appear to be particularly prone to DNA slippage. One can intuitively see how this can happen by looking at Fig. 19.22 . Six genes must work in concert to repair DNA damage. These genes and an example of how we think they work can be seen in Fig. 19.23 .

A paradox of human tumor genesis is that the rates of mutation in normal cells are too low to account for the transformation of benign to malignant. The instability of some colon cancers was the first strong evidence for the so-called mutator phenotype. If a cell cannot repair DNA damage that occurs during the course of normal cell division, mutations can continue to accumulate; eventually, enough mutations occur in genes critical for cell control, and cancer results. Many studies have shown that tumors, which have microsatellite instability (MSI), have defects in mismatch repair. Key metabolic genes usually do not have large areas of microsatellite repeats that are particularly prone to mismatch problems. The mutation rate in nonrepetitive sequences is also elevated 100 to 10,000 times above that in normal cells with normal mismatch repair. Although microsatellite sequences are largely absent from functional genes, mononuclear repeats in some key genes can lead to their inactivation. Examples of such genes include TGF-BR2H (A 10 ), hMSH3 (A 8 ), hMSH6/GTBP (C 8 ), IGF2R (G 8 ), and BAX (G 8 ). Paradoxically, instability of chromosome numbers is often observed in tumors without MMR defects; tumors with MMR defects often have stability of chromosome number.
Mismatch repair proteins interact to form complexes that recognize mismatched DNA base pairs and abnormal loops of DNA. The repair complexes initiated by MMR genes trigger excision and repair of DNA mutations. This system is critically important in ensuring the fidelity of the replicative process and is highly conserved throughout all living organisms. Areas in the genome that contain simple repetitive DNA base sequences called microsatellites (eg, CACACACA or AAAAAAA) are especially prone to mutation. If the MMR system does not function normally, these errors will go unrecognized and continue to accumulate. Cells in which both copies of one of the MMR genes have been inactivated exhibit the so-called mutator phenotype; this allows cells to accumulate mutations as they divide. These random mutations eventually accumulate in a series of genes that are important in controlling cellular growth; the result is loss of growth control and transformation to cancer.
Cells with a defective MMR system exhibit a phenomenon called MSI. This phenomenon occurs as DNA mismatches cause shortening or lengthening of repetitive DNA sequences and these mismatches go unchecked. This results in generation of alleles in the cancer that contain a greater or lesser number of repeats than are present in normal cells from that individual. Laboratory testing for MSI uses an established consensus panel of five microsatellite markers (D2S123, D5S346, D17S250, Bat 25, and Bat 26). Fig. 19.24 shows an example of MSI detection when mismatch prone regions are compared in blood and tumor.

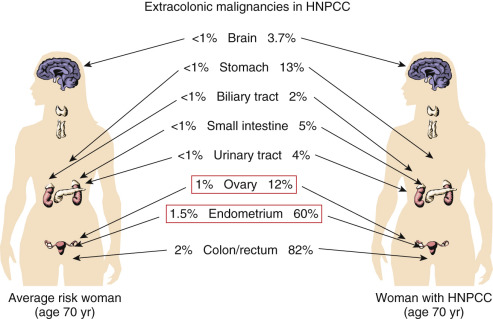
Germline mutations in MMR genes (MLH1, MSH2, MSH6, PMS2) are present in many families and is known as Lynch syndrome. This syndrome is associated with increased risk for colon cancer, endometrial cancer, ovarian cancer, stomach cancer, small bowel, stomach, renal pelvis, biliary tract, and brain cancer. Mutations in one of the Lynch syndrome genes accounts for about 5% of all colon cancers and approximately 2% of all newly diagnosed endometrial cancers.
The Amsterdam II criteria, used to identify Lynch syndrome based on the pattern of cancer in a family is defined as follows:
- 1.
Three or more relatives with Lynch syndrome–associated cancer; one must be a first-degree relative to the other two.
- 2.
At least two successive generations must be involved.
- 3.
At least one of the cancers must have been diagnosed before the age of 50 years.
Seventy percent of patients with that meet Amsterdam II criteria have a germline mutation segregated around an MMR gene: hMLH1 (49%), hMLH2 (45%), and hPMS2 (6%). Approximately 25% of endometrial cancers have an MMR defect; approximately one-fourth of these are germline mutations, and the rest are largely the result of somatic MLH1 inactivation resulting from hypermethylation of the promoter region. MMR will be increasingly investigated and more clearly defined during the next decade.
Telomerase
Normal cells divide; this process is repeated many times as an organism grows and matures. Normal cells stop dividing and terminally differentiate, but cancer cells continue to divide. Cells from younger animals divide more times than do cells from older animals. One difference between normal cells and tumor cells is at the end of their chromosomes. Telomeres, specialized structures at the ends of chromosomes, act as protective caps. In humans, telomeres are made up of 5000 to 15,000 base pairs of TTAGGG repeats. These terminal structures protect the chromosome ends from exonuclease digestion, prevent aberrant chromosome recombination, and form specific complexes that bind proteins.
Normal cells lose about 50 to 100 base pairs from the end of each chromosome every time the cell divides. When a telomere loses a critical number of base pairs, it triggers a signal for the cell to stop dividing and for senescence. Cells have developed several mechanisms to get around this terminal trigger. Some of these are uncommon, such as complex recombination and retrotransport techniques. The most common mechanism is the development of an enzyme complex called telomerase, which adds back telomere sequences lost during replication. Cells that have significant telomerase activity are often immortal cells such as cancer cells and germ cells.
Normal cells lack telomerase activity. Telomerase becomes reactivated in most cancer cells. It is the most prevalent cancer marker known. Therefore, telomerase may be used as a generic cancer marker and as a possible treatment.
Hereditary Cancer Risk Assessment: Genetic Counseling and Genetic Testing
Clinically Relevant Hereditary Syndromes
The advent of linkage analysis and the rapidity of DNA sequencing with other molecular technologies have made it possible to identify many inherited disorders that we could not identify a few short decades ago. Gynecologic malignancies associated with autosomal dominant inherited cancer predisposition that have been identified thus far are as follows:
| Disease | Genetic Abnormality |
|---|---|
| Hereditary breast–ovarian | BRCA-FA pathway genes, including BRCA1, BRCA2, RAD51C, RAD51D, PALB2, BARD1, and BRIP1 |
| Lynch syndrome | MLH1, MSH2, MSH6, PMS2, EPCAM |
| Cowden syndrome | PTEN |
| Li-Fraumeni syndrome | TP53 |
| Ovarian cancer: small cell hypercalcemic | SMARCA4 |
| PPB | DICER1 |
Ovarian cancer
Epithelial
Hereditary breast and ovarian cancer (HBOC): all epithelial histologies; rarely mucinous; most commonly high-grade serous
Lynch syndrome: all epithelial histologies typically endometrioid
Epithelial: borderline tumors
Rarely associated with hereditary cancer syndromes
Germ cell
Not believed to be a part of HBOC
Sex cord–stomal tumors
Granulosa cell tumors
Peutz-Jeghers syndrome (PJS)
Sex cord tumors with annular tubules
PJS
Sertoli-Leydig and juvenile granulosa cell tumors
Pleuropulmonary blastoma (PPB) syndrome
Fallopian tube or primary peritoneal cancer
HBOC
Lynch syndrome
Uterine cancer
Epithelial (majority of uterine cancers)
Lynch syndrome (typically endometrioid, less commonly clear cell; rarely uterine papillary serous (UPSC): not typically thought to be associated with HBOC (reports of UPSC associated with HBOC)
Mesenchymal (endometrial stromal sarcoma, leiomyosarcoma, other nonspecific sarcomas) (rare)
Rarely associated with hereditary cancer syndromes
Uterine soft tissue sarcoma
Li-Fraumeni syndrome
Mixed: Malignant mixed Mullerian tumor (MMMT) or carcinosarcoma, adenosarcoma
Rarely associated with hereditary cancer syndromes; seen in some cases of Lynch syndrome
Cervix
Rarely associated with hereditary cancer syndromes
Cervix adenoma malignum associated with PJS
Vaginal or vulvar cancer
No known association with hereditary cancer syndromes
Gestational trophoblastic disease
No known association with hereditary cancer syndromes
Hereditary cancer syndromes that include increased risk for developing gynecologic cancers.
Commercial clinical genetic testing is available for hereditary cancer risk and has undergone rapid changes in the last several years. Massively parallel sequencing, also called next-generation sequencing (NGS), technology allows for rapid testing of large amounts of DNA sequence. NGS is quicker and less expensive than previous genetic testing tools. It is becoming routine to order a single test that simultaneously evaluates multiple genes thus testing for multiple hereditary cancer syndromes at once. The Supreme Court Decision in June 2013, overturning the ruling on allowing gene patents, particularly BRCA1 and BRCA2 , brought an immediate change in the availability of multigene panel testing through multiple different commercial laboratories.
Our understanding of the tumor spectrum associated with the different hereditary cancer predisposition genes, the risks for cancer, and targeted therapy options is growing exponentially. An increasing number of individuals are seeking the advice of physicians and genetic counselors regarding options for genetic testing hereditary risk for cancer. It is important to have an understanding of hereditary cancer syndromes associated with an increased risk for developing gynecologic cancers.
Hereditary Breast and Ovarian Cancer ( BRCA1 and BRCA2 ; Other DNA Repair Pathway Genes)
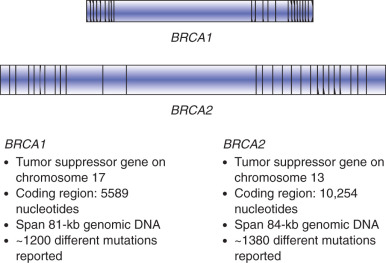
Hereditary breast and ovarian cancer syndrome is the best known and most well characterized of the hereditary cancer syndromes. BRCA1 and BRCA2 and were identified in the early 1990s (1994 for BRCA1 and 1995 for BRCA2 ). Genetic testing for the predisposition to breast and ovarian cancer by testing for mutations in the BRCA1 or BRCA2 genes became clinically available in 1996. The BRCA1 and BRCA2 genes are located on chromosomes 17q and 13q, respectively ( Figs. 19.25 and 19.26 ). Both of these genes are quite large, containing at least 20 exons (coding regions) and are of at least 7000 base pairs (see Fig. 19.26 ). Genetic testing for BRCA1 and BRCA2 has changed significantly since 1996; the initial testing in 1996 only included sequencing for BRCA1 and BRCA2 . This testing was later updated to include five specific BRCA1 rearrangements; deletions or duplications not detected through gene sequencing. In 2006, the addition of deletion and duplication (also called large rearrangement testing) for both BRCA1 and BRCA2 was automatically run for individuals from high-risk families who met certain family history criteria or as an optional add-on for additional cost. Current testing includes sequencing and deletion and duplication analysis automatically for both genes in every test, regardless of personal or family history. Individuals who had genetic testing for BRCA1 and BRCA2 previously may benefit from updated genetic testing, especially if no mutations were identified.
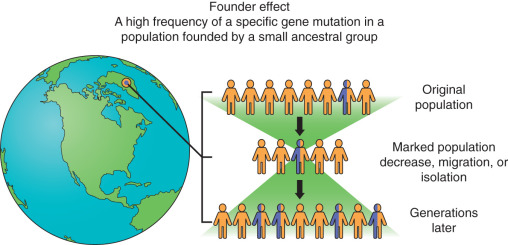
Founder mutations are mutations that are common or even unique to a specific patient population and occur in populations of patients that have been genetically isolated for many generations. The population may have shrunk as a result of a disaster, but the mutation was preserved in a few individuals. When the population reexpands, the frequency of that specific mutation can be quite high within a population ( Fig. 19.27 ). One example of founder mutations occurs in BRCA1 and BRCA2 . ( Fig. 19.28 ). Approximately 2% of individuals of Ashkenazi (Eastern European) Jewish ancestry have one of three founder BRCA mutation: 187delAG ( BRCA1 ), 5385insC ( BRCA1 ), and 6174delT ( BRCA2 ). Historically, targeted testing for the specific found mutations was initiated first with reflex testing to full BRCA1 and BRCA2 gene testing if none of the three mutations were identified. The adoption of NGS tools for genetic testing is changing this process and the specific testing founder mutation testing is skipped and in favor of proceeding directly to multigene testing. These founder mutations are identified with the NGS technology.
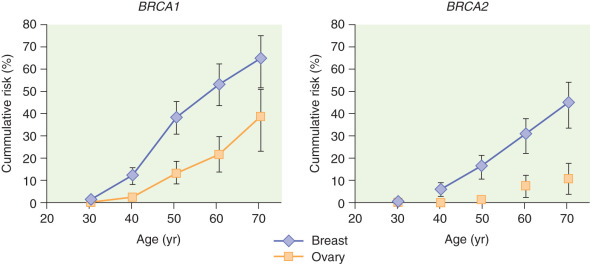
Cancer risks for individuals with BRCA1 or BRCA2 mutation are presented as lifetime risk ranges. These are typically broad risk ranges because the actual reported risk varies depending on the population studied in any given report. Reports of lifetime risk for ovarian cancer ranges from 8% to 62%. The age of onset for ovarian cancer is not necessarily an indication of hereditary risk. Reported lifetime risk for breast cancer range from 41% to 90%. There is limited data to provide individualized risks or mutation specific risks. Unlike ovarian cancer, early age of onset for breast cancer is a hallmark of hereditary risk, but later ages of onset for breast cancer should not be discounted. This should be considered in the context of the cancer family history when determining concern for inherited risk. Additionally, triple-negative breast cancer seems to have a greater association with BRCA1 mutations.
Additional genes in the DNA repair BRCA-FA (Fanconi anemia) pathway were reported to confer increased risk for breast and ovarian cancer beginning around 2010. Genes in the DNA repair BRCA-FA (Fanconi anemia) pathway include BRCA1, BRCA2, CHEK2, BRIP1, RAD50, RAD51C, RAD51D, PALB2, BARD1, MRE11, and NBN. Current information suggests a moderately increased risk for cancer for individuals with mutations in these genes as compared with the higher risks reported with mutations in BRCA1 or BRCA2 . Some of these genes within the BRCA-FA DNA repair pathway may turn out to be associated with a more significant risk for developing cancer than others, and clinical availability of testing this genes will also change over time.
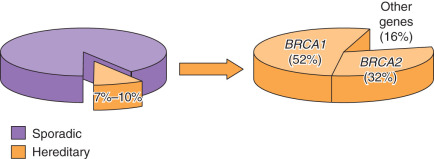
A 2011 study of 360 women with primary ovarian, peritoneal, or fallopian tube carcinoma not selected for age or family history found 24% carried pathogenic mutations in 12 different genes, including BRCA1, BRCA2 BARD1, BRIP1, CHEK2, MRE11A,MSH6, NBN, PALB2, RAD50, RAD51C, and TP53 . The majority were attributed to mutation in BRCA1 or BRCA2 . The remaining portion of inherited risk is due to mutations in one of the Lynch syndrome genes or one of the newly identified BRCA-FA DNA repair pathway genes. The overall contribution of each gene to the risk of ovarian cancer can be seen in Fig. 19.29 . A 2015 study of 1915 women with primary ovarian, peritoneal, or fallopian tube carcinoma not selected for age or family history found 18% carried a pathogenic germline mutation and suggests that 11 genes comprise the majority of hereditary risk for ovarian cancer: BRCA1, BRCA2, BRIP1, RAD51C, RAD51D, PALB2, BARD1, MLH1, MSH2, MSH6, and PMS2 . Mutations in these genes are rare and in some studies not significantly more frequent in ovarian cancer patients compared with control populations so the data continue to emerge to sort out the significance of these and other BRCA/FA pathway genes. National Comprehensive Cancer Network (NCCN) guidelines suggest the following genes to recommend or consider risk-reducing salpingo-oophorectomy (RRSO) if there is a mutation: BRCA1 or BRCA2 and any of the Lynch syndrome genes, BRIP1, RAD51C, and RAD51D . Research continues, and data are quickly evolving regarding the frequency of these mutations associated with specific cancer types, the cumulative risk for cancer, and the most appropriate medical management recommendations for cancer detection or prevention for women with hereditary mutations in these genes.
Cumulative risk is the concept of total risk over time. For individuals with a mutation in a gene involved in a DNA repair pathway, mistakes cannot be adequately repaired that occur with normal DNA replication in cellular division as well as with environmental damage. Therefore, there is a greater risk for developing cancer over time compared with an individual at average risk and the development of cancer tends to occur at earlier ages. Cumulative risks of breast and ovarian cancer associated with mutations in BRCA1 and BRCA2 are shown in Fig. 19.30 .
Pathologic characteristics of tumors have been studied as a method of determining which patients should be referred for hereditary cancer risk assessment. For example, there is a preponderance of triple-negative (ER-/PR-/her2-) breast cancers associated with BRCA1 . High-grade serous ovarian cancer is most commonly associated with mutations in BRCA1 or BRCA2 . However, there are reports of mutations identified in individuals with most of the histologic subtypes. Mucinous ovarian cancers are rarely associated with mutations in BRCA1 or BRCA2 . Current recommendations by the Society of Gynecologic Oncologists and NCCN suggest referral of all patients with epithelial ovarian cancer for genetic counseling and genetic testing.
Identification of germline BRCA mutations, and possibly the other BRCA-FA DNA repair genes, is also becoming more important in treatment decisions. Patients with BRCA1 or BRCA2 mutation–associated cancers tend to have longer survival periods compared with patients with no identifiable germline mutations. These cancers have been found to be more susceptible to chemotherapeutic agents that cause DNA damage. The addition of a poly-ADP-ribose polymerase I (PARP) inhibitor exploits the breakdown of this DNA repair pathway, and these tumors may become even more sensitive to the traditional chemotherapeutic agents. PARP inhibitor Olaparib made by Astra Zeneca is now available for clinical use in patients with DNA repair defects.
Management Guidelines for Hereditary Breast and Ovarian Cancer ( BRCA1 and BRCA2 )
Multiple organizations have published guidelines for genetic risk assessment and management guidelines for individuals with HBOC caused by BRCA1 and BRCA2 with small variances among the different groups. These organizations include the US Preventive Services Task Force (USPSTF), NCCN, American Society of Clinical Oncology (ASCO), European Society for Medical Oncology (ESMO), American College of Obstetricians and Gynecologists (ACOG), and Society of Gynecologic Oncologists (SGO). These guidelines are readily available. The NCCN guidelines are reviewed an updated at least annually. The current recommendations (version 1.2016) suggest clinical breast examination every 6 to 12 months and annual breast magnetic resonance imaging (MRI) beginning at age 25 years. Beginning at age 30 to 75 years, annual mammography and annual breast MRI are recommended, often at staggered 6-month intervals. Women with a BRCA mutation and a personal history of breast cancer should continue with annual mammography and breast MRI. A discussion of the option of risk-reducing mastectomy is also recommended. Data do not support routine ovarian cancer screening. RRSO is recommended between the ages of 35 and 40 years or when childbearing is complete. The option of delaying RRSO until age 40 to 45 years in women with BRCA2 mutations may be considered because there appears to be a later average age of onset of approximately 8 to 10 years compared with BRCA1 mutation carriers. It is important to review a multigeneration family history and take note of the ages of onset of ovarian cancer in the family and adjust this recommendation if there are women with early onset disease. NCCN guidelines also review cancer risks and recommendations for men relative to prostate cancer and mention no specific screening guidelines for pancreatic cancer or melanoma.
Women with BRCA2 mutations of reproductive age should be made aware of options for prenatal diagnosis and preimplantation genetic diagnosis. Cancer family history should be obtained for their partners and consider genetic testing for BRCA2 , especially if there is Ashkenazi Jewish ancestry or family history of breast and ovarian cancer. If both parents carry a BRCA2 mutation, there is risk for a rare autosomal recessive disorder called Fanconi anemia.
As survival improves for patients with ovarian cancer, identification of a mutation can guide cancer screening or prevention recommendations to hopefully keep patients from developing another primary cancer or at least detect the cancer early.
Lynch Syndrome (Hereditary Nonpolyposis Colon Cancer)
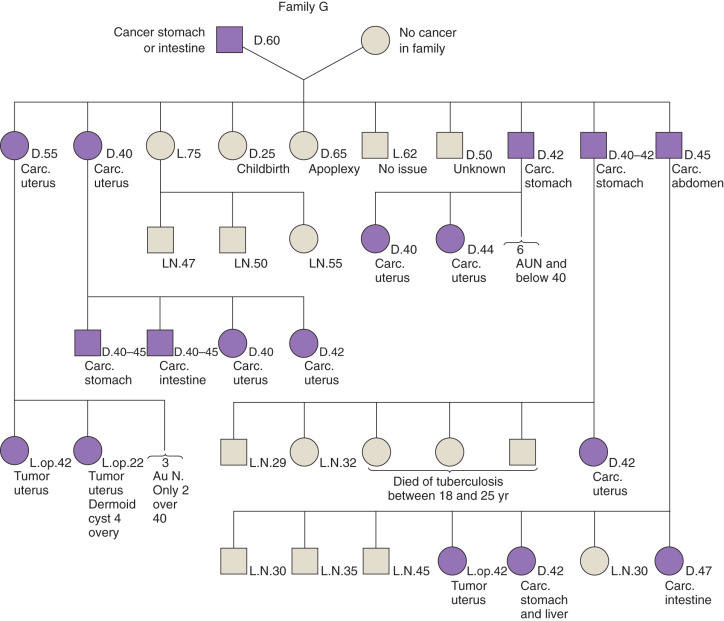
In 1895, a pathologist at the University of Michigan, Aldred Scott Warthin, had a seamstress who had a family with a preponderance of gastric and endometrial cancers. This family became known as family G and is part of the basis for the discovery of Lynch syndrome or HNPCC. Warthin described this family in a report in the Archives of Internal Medicine in 1913 ( Fig. 19.31 ). The family pedigree was updated in 1925 but was largely forgotten until Henry Lynch and colleagues discovered two other families with similar pedigrees in Iowa and Nebraska, which they reported in the 1970s. This was proposed as the “cancer family syndrome” (CFS) in 1971, later changed to HNPCC. The current terminology refers to this syndrome as Lynch syndrome, believing that HNPCC fails to adequately acknowledge the importance of extracolonic malignancies such as endometrial, ovarian, upper gastrointestinal (GI) tract, and urinary tract cancers.