General Principles of Pharmacology: Introduction
The use of drug therapy in elderly individuals has increased substantially in recent years driven by increased numbers of older adults, the results of clinical trials including older adults, and a less nihilistic approach to therapy in this age group. As a consequence, the majority of drug therapy is prescribed to older adults. An understanding of the principles of pharmacology and appropriate application to the individual older person is necessary for all those who develop, regulate, and prescribe and monitor drug therapies in older adults. A further challenge is the need to optimally individualise drug therapy in older adults who constitute a very heterogeneous group ranging from healthy, fit community dwelling individuals taking no regular medication to frail institutionalized individuals with multiple comorbidities and polypharmacy.
When prescribing for older patients, it is important to consider pharmacokinetic and pharmacodynamic changes observed in normal aging, the likely effects of the individual’s genetics and intercurrent disease, as well as evidence for therapeutic efficacy, and safety and the patient’s total exposure to medications. In this chapter, we begin by describing the changes in pharmacokinetics and pharmacodynamics associated with aging and frailty. We then discuss adverse drug reactions and relevant issues in drug development, regulation, and pharmacoeconomics.
Pharmacokinetics
Pharmacokinetic processes determine the relationship between drug input (dose, dosage form, frequency, route of administration) and the concentration of drug achieved over time. The major components of pharmacokinetics are bioavailability, distribution, and clearance. While changes in many of these parameters have been described with aging, the most consistent and marked change in pharmacokinetics in older adults is the increase in interindividual variability.
Bioavailability (F) is the proportion of drug reaching the systemic circulation after administration. Bioavailability depends on the route of administration, the chemical properties of the drug, the absorption of the drug, and the amount of drug that is cleared (first pass loss) before reaching the systemic circulation. Bioavailability of drugs administered by the intravascular route is 100% by definition, and is not affected by aging. Factors that influence bioavailability of medications for common extravascular routes of administration, and any known changes that occur with aging, are shown in Table 8-1. Bioavailability is the ratio of the area under the concentration–time curve (AUC) when the drug is given extravascularly to that when it is given intravenously (Figure 8-1). Bioavailability (F) is expressed as a fraction or percentage:
The bioavailability of a drug given extravascularly can vary with age. As shown in Figure 8-1, with aging, absorption is often slower so the maximal plasma concentration is reached later (longer Tmax) and is lower (lower Cmax). However, the extent of absorption is usually complete in older adults so the area under the curve (bioavailability) is not affected. Drugs that require an acidic environment for absorption, such as ketoconazole, ampicillin, and iron, may have a reduced extent of absorption in the 5% to 10% of older adults with age-related hypochlorhydria secondary to atrophic gastritis, and in those taking medications that raise gastric pH such as H2-antagonists and proton pump inhibitors. In the case of oral administration where the medication undergoes first pass metabolism, which is often decreased in older adults, the area under the curve (bioavailability) may be increased.
ABSORPTION | FIRST PASS CLEARANCE | ||||
|---|---|---|---|---|---|
ROUTE | PROPERTIES OF DRUG | Description | Age-Related Changes | Description | Age-Related Changes |
Oral | Particle size formulation Lipid solubility Ionization | Gut lumen decomposition | Gastric pH may be less acidic, altering ionisation | Gut wall CYP450 metabolism | Not known |
Passive absorption | Complete but slower – Increased by longer gastrointestinal transit time and possible increase in permeability of epithelium – Decreased by reduced perfusion | Hepatic metabolism | Reduced 30–50% | ||
Active transport (e.g., iron, vitamin B-12) | Probably decreased | ||||
Gut wall P-glycoprotein transports drugs back into lumen | Not known | ||||
Sublingual | Particle size Lipid solubility Potency | Rapid into blood vessels at base of tongue | Not known ?Reduced perfusion | Nil | |
Rectal (local and systemic action) | Lipid solubility Ionisation | Varies with rectal contents | Not known ?Reduced perfusion | Nil | |
Subcutaneous Injection | Particle size (small particles absorbed by capillaries, large particles absorbed by lymphatics) Protein complex pH Use of vasoconstrictors | Slow | Not known ?Reduced perfusion ?Changes to lymphatics | Nil ?Proteolysis of protein drugs in lymph nodes | |
Intramuscular Injection | Lipid solubility Particle size (small particles absorbed by capillaries, large particles absorbed by lymphatics) | Slow (faster than subcutaneous due to better perfusion) | Not known ?Reduced perfusion ?Changes to lymphatics | Nil ?Proteolysis of protein drugs in lymph nodes | |
Percutaneous | Lipid solubility | Slow Heat dependent | Not known ?Reduced perfusion | Nil | |
Intranasal (local and systemic) | Lipid solubility | Variable | Not known ?Reduced perfusion | Nil | |
Inhaled (local and systemic) | Particle size – Powders – Aerosol solutions inhaler (type and how used) Gases: gas partition coefficient (blood) | Minimal systemic absorption | Not known ?Effects of reduced alveolar area, low-grade inflammation, ventilation/perfusion mismatch, decreased diffusion and transport across alveolar capillary membrane | Lung metabolism and clearance | Not known |
Ophthalmic (topical) | Formulation: drops, suspensions, ointments | Minimal (drainage through nasolacrimal canal) | Not known | Nil | |
Figure 8-1.
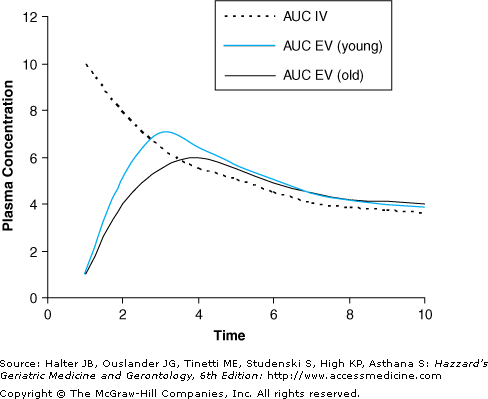
Theoretical plasma concentration–time curves for the same drug given intravascularly (IV) and extravascularly (EV). The bioavailability is the ratio of the area under the curve (AUC) for the EV route to the AUC for the IV route. The intravascular AUC is not affected by age. With aging, the extravascular curve shows delayed and lower maximal plasma concentration and AUC may be increased for drugs that undergo first pass hepatic metabolism.
Bioavailability defines the dose adjustment between intravascular and extravascular drug administration. Lower bioavailability drugs require higher doses when given extravascularly than intravascularly. Reduced first pass hepatic metabolism in older adults results in a lower dose requirement for oral active drugs that undergo hepatic clearance. Examples of prodrugs, which are administered as inactive compounds that must be metabolized into the active drug, include codeine, propranolol, enalopril, perindopril, and simvastatin. In older adults, there may be a higher dose requirement for prodrugs to obtain the same AUC for the active drug (metabolite) as in a young person.
The effects of aging on bioavailability of modified release oral formulations are not known. Modified release oral formulations tend be released for absorption lower in the gastrointestinal tract. In young adults, cytochrome P450 3A4 (CYP 3A4) has the highest content in the duodenum, followed by the jejunum, ileum, and colon. Drugs released lower in the gastrointestinal tract undergo less gut wall metabolism, increasing their bioavailability. Expression of CYP 3A4 in the gut wall has not been studied in aging. The activity of the transporter, P-glycoprotein (P-gp), which pumps drugs from the enterocytes back into the gut lumen, has its highest concentration in the ileum, followed by the colon, jejunum, and duodenum. Therefore, modified release formulations, released lower in the gastrointestinal tract, may undergo less P-gp efflux back into the gut lumen, increasing their absorption. There is no change in expression of P-gp in intestinal microsomes in aged rats; it is not known how intestinal wall transporter activity varies with age in humans. The combination of modified release medications, the possibility of less gut wall metabolism lower in the gastrointestinal tract and slower transit time with aging may increase drug bioavailability in older adults, although the different formulations of modified release medications may be differentially affected by age. Some formulations rely on pH to dissolve an external coat and may be affected by higher gastrointestinal pH in some older adults. Others have a constant drug delivery rate independent of pH or gastrointestinal motility and may be less affected by changes of aging.
Volume of distribution (Vd) is the apparent volume that the drug distributes into to achieve the plasma concentration (Cp). Vd is not a real anatomical volume. Vd may exceed the total real volume of the human body when there is significant binding to tissues. Vd is expressed by the equation:
The amount of drug in the body is known immediately after a known dose of the drug has been given intravenously, before it can be eliminated. Therefore, Vd can be calculated in humans from the dose given intravenously and the Cp immediately after administration.
The volume of distribution of a drug depends on plasma protein binding, lipid to water partition coefficient, tissue binding properties, and transporters. The main determinant of volume of distribution is the ratio of the strength of binding to plasma proteins to the strength of binding in tissues. Drugs that are bound more strongly to plasma proteins have a smaller Vd, and those that are bound more strongly to tissues have a larger Vd. Some examples include heparin and warfarin, which are highly plasma protein bound, and have Vd of 5 and 8 L, respectively, similar to blood volume. Digoxin is water-soluble but highly bound to muscle, with a Vd of 15 L. Imipramine is lipid soluble and has a Vd of 2100 L, which is far greater than the 42 L real volume of a 70 kg human. Alendronate adheres strongly to the bone surface, and has a Vd of 2580 L. The major determinants of Vd and how they are affected by normal aging are shown in Table 8-2.
FACTOR | EFFECT ON Vd | AGE-RELATED CHANGES | CLINICAL APPLICATION |
|---|---|---|---|
Plasma protein binding | Highly protein bound drugs are generally less able to cross membranes and have smaller Vd | Decreased albumin which binds acidic drugs, e.g., warfarin, NSAIDs, phenytoin. Increased α1-acid glycoprotein which binds basic drugs, e.g., verapamil, propranolol. | Not usually clinically significant. |
Tissue binding properties | Drugs which are tightly bound to tissues have large Vd | Changes in body composition (sarcopenia, increased adiposity) may affect Vd. | Drugs bound to muscle, e.g., digoxin, have decreased Vd with aging. Therefore decreased loading dose required. |
Lipid: water coefficient | Lipid soluble drugs can pass through lipid membranes of cells more easily and have higher Vd than water soluble drugs | Relative increase in proportion of body fat and decrease in body water (muscle mass). Therefore, higher Vd for lipid soluble drugs and lower Vd for water soluble drugs with aging. | Loading dose of water soluble drugs, e.g., gentamicin, digoxin, decreased with aging to avoid toxicity from high initial Cp. |
Transporters | Passive facilitated diffusion (move drug in same direction as concentration gradient). Active transport (use ATP to move drug against concentration gradient). | Unknown. | Drug interactions may occur at level of transporters. Passive transporters include the Organic Anion Transport Proteins (OAT-P) for benzyl penicillin, digoxin, and pravastatin. Active transporters include MDR1 (P-gp) for many cationic or neutral drugs, e.g., digoxin, macrolide antibiotics, verapamil. |
The immediate volume of distribution is blood volume. Then the drug is distributed from the blood into various tissues at a rate that depends on the perfusion of the tissue, the ease with which the drug can pass through lipid membranes of cells and any active transport. Membrane permeability and passive transport may increase with age and malnutrition. The effects of aging on active transport is not well documented in humans, and in rats, varies between tissues, with an increase in P-glycoprotein expression and activity in the liver and lymphocytes, but not in the intestine or endothelium, with aging. With aging, the volume of distribution of verapamil (a substrate of P-glycoprotein) in the brain increases, consistent with dysfunction of the blood–brain barrier. Tissue perfusion may decrease with aging, which may slow distribution, particularly to less highly perfused tissues such as muscle and fat. The rate of distribution to or from the site of action of a drug may determine the onset or offset of drug effects.
The volume of distribution determines the loading dose of a drug. The loading dose is the dose required to achieve the desired Cp as soon as possible, and is calculated with the equation:
There may be small changes in loading dose in older adults due to the changes in Vd described above. For example, the volume of distribution of digoxin decreases from approximately 7 to 6 L/kg with aging, probably due to reduced muscle mass and reduced binding of digoxin to muscle. Therefore, loading dose should decrease by approximately 15%.
Clearance is the rate of elimination of a drug from the body. Clearance, CL, from each organ of elimination can be defined by the equation:
where Q is the flow rate to the organ and E is the extraction ratio. Total body clearance is the sum of the drug clearances by each organ, and can be expressed as
Clinically, the clearance affects the maintenance dose of medications.
Removal of a drug by the liver depends on portal and arterial hepatic blood flow and elimination by metabolism and/or secretion into the bile. The processes involved in hepatic clearance are shown in Figure 8-2 and changes in normal aging are shown in Table 8-3. Hepatic clearance of highly extracted substrates is determined mostly by hepatic blood flow and is known as flow-limited metabolism. This is consistently reduced in older adults. Hepatic clearance of poorly extracted substrates is described as capacity-limited metabolism because the intrinsic clearance (metabolising capacity) is the rate-limiting step and is less affected by old age. Protein binding may also affect the clearance of poorly extracted substrates. The effects of normal aging on flow-limited and capacity-limited hepatic clearance are shown in Figure 8-3.
Figure 8-2.
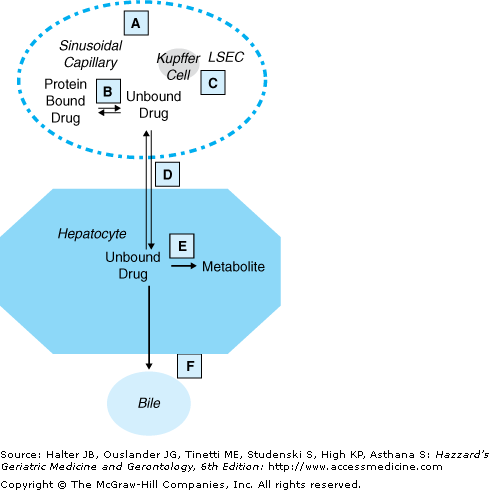
The processes involved in hepatic clearance. A, blood flow; B, protein binding; C, scavenger cells (Kupffer and Liver sinusoidal endothelial cells, LSECs); D, transfer across the LSECs and the hepatocyte apical membrane; E, metabolism; F, transport into bile ductule. Adapted from Tozer TN, Rowland M. Introduction to Pharmacokinetics and Pharmacodynamics: The Quantitative Basis of Drug Therapy. Lippincott Williams and Wilkins; 2006, Fig 5–17.
PROCESS OF HEPATIC CLEARANCE | DESCRIPTION | AGE-RELATED CHANGES | CLINICAL APPLICATION |
|---|---|---|---|
Hepatic blood flow | Portal venous flow (∼80%) and hepatic arterial flow (∼20%) | Decreases by 30–50% | Reduced clearance by 30–50% of high extraction ratio drugs, e.g., morphine and verapamil. Less impact on low extraction ratio drugs, e.g., carbamazepine, warfarin, diazepam. |
Protein binding | Only free drug is cleared. Protein binding affected by disease and competition from other drugs. | Decreased albumin: acidic drugs have higher fraction unbound and increased hepatic clearance. Increased α1-acid glycoprotein: basic drugs have lower fraction unbound and decreased hepatic clearance. | Only significant for drugs that are highly protein bound (>90%) with low hepatic extraction ratios, e.g., warfarin, phenytoin, diazepam. |
Scavenger cells | Kupffer cells scavenge large protein drugs. Liver sinusoidal endothelial cells (LSECs) may scavenge smaller particles. | Possible reduction in scavenger function (in animal studies). | Not known. May reduce hepatic clearance. |
Transfer into hepatocyte | Transfer across the LSECs and the hepatocyte apical membrane by passive or active transport. | Structural changes in LSECs and the space of Disse may reduce transfer. Changes in hepatocyte membrane transport not known. | Not known. May reduce hepatic clearance. |
Metabolism | Biotransformation to more hydrophilic metabolites that may be equally, less or more active than parent drug. | Reduced phase I metabolism in vivo in normal aging by 30–50%. Phase II metabolism appears to be maintained in healthy aging, reduced in frail aged. | Probably preserved clearance of drugs that undergo capacity-limited phase II metabolism in healthy aging, e.g., temazepam, salicylic acid. |
Transfer into bile ductule | Active transport into bile canaliculi. Bile enters small intestine and drug or metabolite reabsorbed (enterohepatic cycle) or excreted in faeces. | Unknown. In aged rodents, increased biliary P-glycoprotein expression and function. | Unknown. |
Figure 8-3.
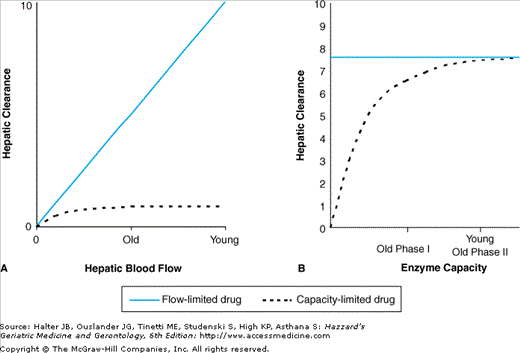
The effects of normal aging on hepatic drug clearance. With aging, hepatic blood flow is reduced by approximately 50%, which is associated with a 50% reduction in clearance of flow-limited drugs, but has little effect on clearance of capacity-limited drugs (A). In normal aging, phase I metabolism is reduced, which reduces hepatic clearance of capacity-limited drugs metabolized by these enzymes, while phase II metabolism is probably preserved, although it may be reduced in frailty. Changes in enzyme capacity do not affect the clearance of flow-limited drugs (B).
Normal aging is only one of many factors that may influence hepatic clearance. For example, the hepatic clearance of flow-limited drugs may be decreased by diseases that reduce hepatic blood flow, such as heart failure, while hepatic clearance of capacity-limited drugs may be altered by other drugs that induce or block hepatic enzymes or by genetic polymorphisms.
The majority of drug elimination occurs through the kidneys. In the kidneys, drugs and their metabolites may undergo glomerular filtration, tubular secretion, and tubular reabsorption. These processes are summarized, with descriptions of any known age-related changes and relevant examples in Table 8-4.
PROCESS OF RENAL CLEARANCE | EFFECTS OF AGE | CLINICAL APPLICATION | EXAMPLE |
|---|---|---|---|
Glomerular filtration | Decreased GFR, extent unclear, ∼10–40% | Estimates used to adjust maintenance doses of drugs for renal impairment | Gentamicin clearance correlates with Cockcroft-Gault estimates of creatinine clearance. |
Tubular secretion (active) | Decreased | Reduction in renal clearance may be greater than reduction in GFR. With polypharmacy, increase risk of drug-drug interactions through competition for transporters. | Ratio of procainamide clearance to creatinine clearance decreases with aging. Digoxin excreted by passive glomerular filtration and active tubular secretion. Serum digoxin levels increase with number of concurrent P-gp inhibitors, e.g., verapamil, erythromycin, amiodarone, spironolactone, atorvastatin. |
Tubular reabsorption (passive) | Unknown | If impaired, would reduce the effect of reduced glomerular filtration on clearance. | Changes in clearance of lithium, which, like sodium, is freely filtered at glomerulus and 80% reabsorbed in the proximal tubule, consistent with changes in GFR with aging. |
At the glomerulus, drugs and their metabolites are passively filtered from the afferent arterioles through the glomerular membrane to produce an ultrafiltrate of plasma within the capsular space of the glomerulus. Plasma proteins and cells are too large to cross the glomerular membrane so only free drug enters the capsular space of the glomerulus. The glomerular filtration rate (GFR) is the total volume of glomerular filtrate produced per unit time by all nephrons, and is approximately 120 mL/min in a young, healthy person (∼10% of total renal blood flow). In normal aging, recent estimates suggest that GFR is moderately reduced by 15% to 40%. Effective renal plasma flow (ERPF) decreases proportionally more than GFR, by ∼10% per decade from young adulthood to the age of 80 years. Older adults have increased renal vascular resistance, with impaired relaxation in response to vasodilators.
Estimation of renal function in older adults is complicated by confounders. Serum creatinine is not useful because creatinine production is reduced in older adults, due to decreased muscle mass. The Cockcroft-Gault equation [6], which includes age, weight, and serum creatinine, is used clinically to estimate creatinine clearance:
Most dosing guidelines use the Cockcroft-Gault equation to adjust doses of renally excreted drugs with narrow therapeutic indices, such as gentamicin and low-molecular weight heparin, for renal function. However, the Cockcroft-Gault equation was derived from observational data from males receiving routine medical care for a range of illnesses, and may underestimate renal function in normal aging. Much of the age-related decline in renal function may be related to disease, particularly hypertension, atherosclerosis, and heart failure, rather than normal aging.
Recently, an equation to estimate the GFR, the modification of diet in renal disease (MDRD) equation, has been introduced to clinical practice. This equation has not been validated for extremes of age or dose adjustment. Unadjusted for body surface area, in the presence of the reduced height and weight observed in normal aging, the MDRD is likely to overestimate renal clearance in older adults. A study of elderly patients in long-term care demonstrated that only one-third of patients were classified in the same stage of renal impairment by both methods, with average MDRD estimates 20 mL/min/1.73 m2 higher than those determined using Cockcroft-Gault, suggesting that the methods are not interchangeable for determining drug dose. Further validation studies are required to determine the best method for clinically determining renal impairment for the purposes of adjusting drug doses. Recently, it has been suggested that renal function in older adults could be better estimated using methods independent of serum creatinine, such as Cystatin C, an endogenous polypeptide marker of renal function, which is significantly elevated in older adults. The role of this marker in clinical practice is under investigation.
Currently available methods to estimate renal function in older adults are not reliable, particularly in frail or acutely ill older adults. While the Cockcroft-Gault equation can give an estimate of renal function for dose adjustment of renally excreted drugs, the maintenance dose of drugs with narrow therapeutic indices should be guided by therapeutic drug monitoring and clinical response.
Drugs may also enter the renal tubules through active secretion. The unfiltered fraction of plasma and the particles too large to enter the glomerular filtrate pass through the efferent arterioles to the vessels supplying the renal tubules. The majority of tubular secretion occurs in the proximal convoluted tubule and relies on active transport by specific pumps for anions and cations. Drugs bound to proteins or cells can undergo tubular secretion, but not glomerular filtration. These pumps are saturable and drugs may compete for transport into the renal tubule. There is a reduction in tubular secretion with aging to a similar and possibly greater extent than GFR.
Drugs that have been filtered at the glomerulus or secreted in the proximal convoluted tubule may be passively reabsorbed down the concentration gradient from the tubular fluid in the proximal and distal tubules. Reabsorption occurs for lipid-soluble drugs that are not ionized, and therefore may be influenced by the pH of the tubular fluid. Reabsorption is inversely related to urine flow rate. Changes in tubular reabsorption with aging are not well described. Proximal tubule functions are generally preserved in healthy aging, with near normal production of erythropoietin and normal sodium reabsorption in the proximal tubules. However, overall tubular function is decreased in the elderly, with impaired ability to concentrate or dilute urine maximally.
Drug metabolism through cytochrome P450 and via conjugation can occur at many sites outside the liver. The effect of intestinal cytochrome P450 on bioavailability has been discussed in the section on bioavailability and in Table 8-1. Proteases and peptidases are found throughout the body and metabolise polypeptide drugs such as insulin, erythropoietin, and interferon. Circulating esterases also play a role in drug metabolism, including acetyl cholinesterase and carbonic anhydrase. Age-related changes in clearance outside the liver and kidney are not known.
Half-life (t1/2) is the time taken to halve the amount of drug in the body (or the plasma concentration of the drug). Half-life depends on volume of distribution (Vd) and clearance (CL), and is calculated using the following equation:
The constant, 0.693, is the natural logarithm of 0.5.
A decrease in clearance, which is seen commonly with aging, results in an increase in half-life. An increase in volume of distribution, which occurs with lipid-soluble drugs with aging, would also result in an increase in half-life. Higher volume of distribution indicates that the drug is more concentrated in the tissues than the blood. Hepatic and renal clearance act on drug in the blood, so a smaller proportion of drug is eliminated with a larger volume of distribution. The theoretical effects of aging on half-life are shown in Figure 8-4.
Figure 8-4.
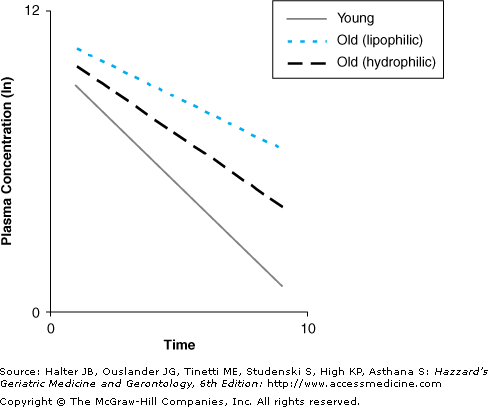
The theoretical effects of aging on half-life. The decrease in clearance with aging prolongs half-life. For lipid-soluble drugs, the increase in volume of distribution prolongs half-life further. For water-soluble drugs, the decrease in volume of distribution reduces half-life, but in most cases, the size of the effect of aging on volume of distribution is smaller than the effect on clearance, resulting in an overall increase in half-life compared to young subjects.
The half-life determines the time course of drug accumulation and elimination, and the choice of dose interval. When a drug is started, plasma concentration reaches >90% of steady state after approximately four half-lives. When a drug that was at steady state is stopped, plasma concentration reaches <10% of steady state after approximately four half-lives. Dose interval is usually similar to half-life to maintain steady state.
Changes in half-life with age will affect how long the drug takes to accumulate, to be eliminated, and the dosing interval of the drug. The increase in half-life that is observed for many drugs with aging means that it will take longer for drugs to reach steady state, longer to be eliminated after they are ceased, and dosing intervals may need to be increased.